0. foreword
最近看了一篇比较有意思的文章,而且要讲组会了,认真学习一下顺便做个随笔当做我讲组会的草稿
(文章并不是直接翻译,文章的内容按照自己的理解进行了些改动)
1. Abstract
变分自编码器是一种无监督的生成模型,当把它应用在蛋白质数据上的时候,可以利用它按照系统发育关系对蛋白质进行划分,并且可以利用它生成新的序列,这些序列保留了蛋白质的内在统计特征.
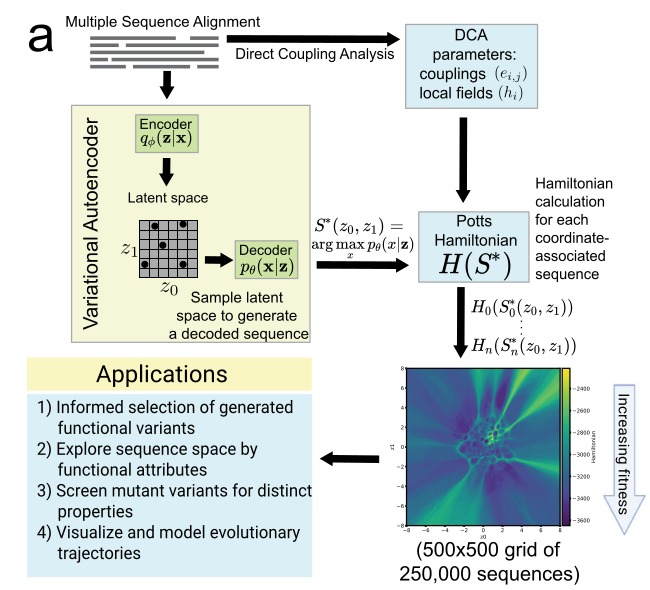
之前的一些研究主要集中于蛋白质序列的聚类和生成的蛋白质的特征,在这里作者评估了蛋白质序列所嵌入的隐空间的流形.为了探索隐空间流形,作者使用变分自编码器和potts model来构建潜在生成景观 (Latent Generative Landscapes,LGL)
作者展示了这样一个景观用于解析蛋白质系统发育关系,功能以及适合度特征,作为例子作者探索的蛋白质家族包括珠蛋白家族,beta内酰胺酶,离子通道蛋白和转录因子等.
同时作者利用实验数据表明这样一种地形图是有助于我们理解序列多样性和蛋白质进化的.
2. Introduction
2.1 background
本文的标题可以译作“把潜在生成景观作为蛋白质功能多样性的地图”(有点僵硬)
我们知道在蛋白质演化过程中所经历的突变,比如氨基酸的替换,缺失,插入,序列的重组等等,都会受到蛋白质功能,结构稳定性等因素的约束作用,也就是自然选择. 这种约束作用导致只有某些特定的突变才能在种群中被固定下来,这造就了现今观察到的执行特定功能的蛋白质家族内部都会存在特殊的统计信号,通过解析这些统计信号人们可以对蛋白质中重要的相互作用和位点进行探索.
用于解析这些统计信号的方法有很多,有一些机理模型如EVcoupling,DCA(Direct Coupling Analysis),这些工具在过往的研究当中被用于蛋白质内部位点的接触预测,以及蛋白质之间的作用位点的预测.像作者在文中所列举的方法都是基于potts model的方法,它仅仅需要你向这个模型中输入蛋白质的多序列比对就可以了,实际应用的时候这些多序列比对的规模会很大通常是数千条的样子,同时要求序列的同质性不要太高.
还有一些工具就是利用机器学习的方法,比如alphafold,它可以对蛋白质的结构进行预测,deepppi可以用来研究蛋白质的相互作用,RBM受限玻尔兹曼机可以用于检测特定的motif(与特定功能相关的序列基序),还有变分自编码器VAE可以用于蛋白质序列的分类以及突变效应的预测.
在机器学习的方法中,有很多方法是监督学习的范式,需要我们在给出序列的同时给出序列对应的标签,比如蛋白质的结构数据,蛋白质的适合度这些都是要实验测定的,规模很难达到我们在数据库中所能得到的序列数据的水平,如果没有大量的数据这些方法的应用会受到限制.
而无监督学习的范式相对而言就方便了许多,但是很多信息是隐藏起来的,不像监督学习是端到端的,以蛋白质和vae为例,vae作为一种无监督学习方法,它的降维能力可以在低维空间上很好反映数据在原始序列空间中的分布情况,但是它的分布所展现出的功能意义,我们需要对模型,或者对embedding的分布进行进一步的调查,简单的比如对训练数据进行聚类,复杂的会对模型本身进行分析.
总结一下,监督学习研究蛋白质的思路很简单,但是获取"标签"的工作量很大;无监督学习研究蛋白质思路会复杂很多,但是不需要标签,有得有失.
2.2 VAE and Potts model
在这篇文章中变分自编码器和potts model得到了作者的注意,对于变分自编码器和potts model可以单拿出两个小节来讲或者再出两个随笔.
2.2.1 VAE(Variational Autoencoder)
简单来说变分自编码器是一种无监督的生成模型,它可以帮助我们对数据进行降维,和GAN(生成对抗网络)一样能生成具有某种特征的数据(比如图片,文字,在本文中就是蛋白质序列)
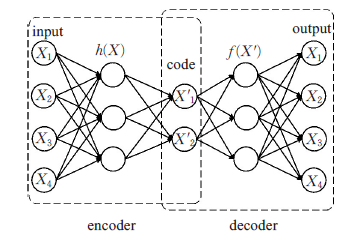
我们想要理解VAE的设计,需要先要了解自编码器AE(AutoEncoder)
AE也是一种无监督的学习模型,它具有两个主要的结构一个是编码器,一个是解码器

(这是一张蛋白质家族的AE嵌入分布图引自Learning meaningful representations of protein sequences)
编码器负责将一个输入数据映射到一个数值编码,也就是将输入数据所在的原始空间变换成一个潜在空间
解码器负责将潜在空间中的数值编码映射为原始数据空间中的一个点,也就是将潜在空间变换回原始空间
在模型的训练过程中编码器将一个输入数据A转变为一个数值编码,数值编码紧接着就输入到解码器中输出新的数据A',损失函数要求输出的数据A'要尽可能的像输入数据A.所以总的来说训练过程中的AE将编码器和解码器作为一个整体,将一个信号输入这个黑箱,黑箱要能很好的还原这个信号.
这样训练的目的就是希望低维的潜在空间中的点能够真正的代表高维原始空间中的数据,通过低维空间上的分布近似反映数据在高维空间上的分布,经过降维排除一些无用的特征,保留重要的信息可以便于下游的应用,计算和可视化.
相比t-SNE,PCA这些降维方法可以更好的识别数据的特征,PCA他是线性的,对非线性的特征识别能力不行,而t-SNE需要提前确定一种度量来衡量两个数据点之间的距离,比如我们衡量两个蛋白质序列的进化距离,我们先验的认为不同位点的进化速率相同,都遵循同一种氨基酸替换规则,而真实情况要复杂的多,导致这种度量并不合理,不同的度量得到数据的t-SNE嵌入,数据的分布情况可能并不一直,而AE不需要这些先验的认识.
可是AE也有它自己的缺点,AE的训练容易导致过拟合,使得潜在空间中的点并不能很好的表示原始信号中的内涵,也就很难利用它的解码器生成一些数据,举一个博客上给出的例子,假如有一个自编码器,它的潜在空间只有一个维度,然后在潜在空间中数字1表示"新月",数字10表示"满月",你可能会想数字5会不会代表半月,但是自编码器在训练的时候只见过新月和满月,并不清楚应该让5代表什么.真正的半月可能由数字100来表示了,以上的说法只是打比方并不严谨.

我们可能希望5来代表半月,这样更符合我们对月亮随时间阴晴圆缺连续变化的认识,我们希望模型也能展现这样一种情景,一种处理方法就是在编码器中我们不让原始空间的点直接映射到嵌入空间的点上而是让它映射到某一个多元正态分布上,这就是VAE做的事情.
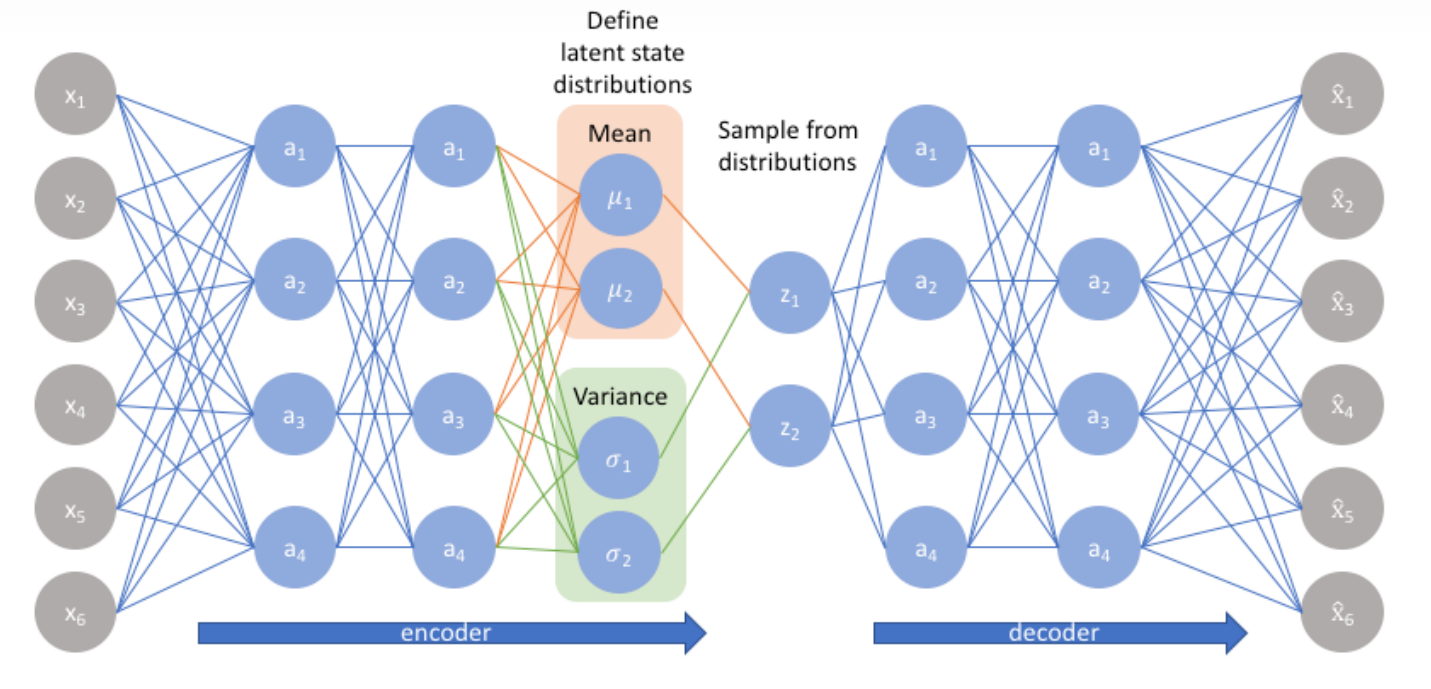

假如我们让新月映射到均值为1的正态分布上,让满月映射到均值为10的正态分布上,这样潜在空间上每个点都可能是新月只不过在1那个点上最像,越向1靠拢概率越高,同样的每个点都可能是满月只不过在10那个点上最像,越向10靠拢概率越高.这样一来,假如两个正态分布的方差是合适的5这个点既像满月也像新月,同时具有二者的特征那生成出来很有可能是半月了.

之前说的AE嵌入分布图,把架构改成VAE之后的嵌入分布
所以总的来说VAE是AE的plus版,使得这种基于编码器和解码器的架构既能用于降维也能用于生成数据.在这篇文章中作者通过一个潜在空间近似的表征蛋白质的序列空间.
最后说一下把蛋白质训练数据是如何输入VAE模型和VAE的具体架构,我们知道构成蛋白质序列的基本氨基酸有20种算上硒代半胱氨酸和吡咯赖氨酸和多序列比对产生的gap一共23种字符,我们对已经对齐的序列集中的每条序列的每个氨基酸进行one-hot编码,这样每个序列可以有23*序列长度的矩阵来表示,矩阵reshape成一个向量(向量长度23*序列长度)这就是VAE真正的输入.它经过编码器的两个全连接层最终生成多元高斯分布的均值和标准差,在本文中高斯分布为两个维度,解码器接受一个二维向量通过两个全连接层借助softmax函数最终返回每个位点上23种字符出现的多项分布.它的损失函数是elbo.
Extra:ELBO
推荐阅读:
An Introduction to Variational Autoencoders
Auto-Encoding Variational Bayes
我们假设我们的蛋白质序列集中各个序列是独立同分布的(由于我们数据集中的天然序列基本都源于一个共同祖先所以这个假设并不正确,但是为了计算可行性,我们需要做出妥协),来自于一个未知的分布\(p_{\theta}(x)\)这个分布非常的复杂
通过全概率公式我们能够知道
我们假设我们的数据来源一种包含隐变量z的生成过程,\(p_{\theta}(z)\)是一个标准正态分布,我们假定真正的\(p_{\theta}(x|z)\)的形式是一个decoder.
另一方面,对于真实的后验分布\(p_{\theta}(z|x)\)我们并不知道或者说太难用解析的方法推导出来,所以我们使用一个encoder \(q_{\phi}(z|x)\)来代替
我们的蛋白质序列数据集表示为\(X = \{x_{1},..x_{i},....x_{n}\}\),\(x_i\)为数据集中的一个序列

对于这样一个数据集它出现的概率是\(p_{\theta}(X)\)
换一种形式\(\mathcal{L}\):likelihood
对于任意一个函数\(f(x)\)和条件概率分布\(p(z|x)\)都会有
以及对于任意一个概率分布\(p(x,z)\)都会有
所以
其中\(p_{\theta}(x|z)\)是vae中的解码器给出的当前数据出现的概率,\(p_{\theta}(z)\)是标准多元正态分布中z的概率密度,\(q_{\phi}(z|x)\)是编码器所确定的多元正态分布中\(z\)的概率密度,\(x\)是我们的序列数据,\(z\)就是潜在空间中的一个点
由于ELBO是一个积分所以我们采取抽样的方式来代替
于是
更进一步的具体的表达式不太好写这里就不给出了,在模型训练过程中还涉及到重参数化技巧使得反向传播在采样过程也能够顺利进行.
因为\(p_{\theta}(z|x)\)未知,我们并不知道\(\mathcal{L}\)中的KL散度如何计算,作为妥协我们只针对ELBO进行优化,通过优化ELBO提高模型似然的下界,进而提高模型的似然度,通过这种方式让我们的给定分布去逼近真实分布.
2.2.2 Potts Model
推荐阅读:
这篇文章并不是使用potts model研究蛋白质的起源,感兴趣可以自行搜索
然后是potts model,我本人没怎么学过统计物理,所以在这方面不是很懂
它的本质是一种玻尔兹曼模型(玻尔兹曼分布的推导可以自行查看各类统计物理学书籍),它把一个蛋白质家族看作是一个系统,系统中包含有很多粒子,这些粒子就是蛋白质序列,特定的氨基酸排列代表着一类粒子.同种粒子都具有相同的能量(哈密顿能量),他们的能量是由它们的氨基酸排列所决定的.
不同种类的粒子在系统中的含量并不相同,能量水平越高的一类粒子在系统中的含量会更少.对于一个蛋白质家族来说,如果一个蛋白质序列作为该系统中粒子能量很高,那么就代表着这个蛋白质序列不像是具有该蛋白质家族功能和结构的成员.
总而言之,Potts model经过一个蛋白质家族的多序列比对的训练,估计参数之后,将一个蛋白质输入到这个模型中,它可以对应的返回一个能量,来表征这个序列具有该蛋白质家族特性的程度.能量越低说明越具有该蛋白质家族的特性.
在本文中作者将哈密顿能量也看作一种适合度,用它表征一种基因型具备某一类功能或结构的程度,而严格的适合度的定义是一种生物的基因型适应环境并向后传递后代的能力,需要我们注意区分.
Extra potts model的数学形式
\(x\)是一种蛋白质序列,\(x_i\),\(x_j\)代表的是蛋白质\(i\)位点和\(j\)位点上的氨基酸,\(h_i(x_i)\)表示的是当\(i\)位点上的氨基酸为\(x_i\)的时候该为点的哈密顿能量,\(e_{ij}(x_i,x_j)\)表示的是当\(i\),\(j\)位点上的氨基酸类型分别为\(x_i\),\(x_j\)时它之间的相互作用所具有的哈密顿能量,通过对所有位点对和所有单位点能量的加和表征一个序列在该系统下所具有的能量
配分函数Z
该式表示蛋白质序列x在一个蛋白质家族系统中出现的概率
2.3 作者的idea
因为变分自编码器这种无监督学习的特性和生成数据的能力,再加上作者认为potts model能够以哈密顿能量作为蛋白质适合度的一种表示,因此作者考虑将二者结合起来,提出了一个概念LGL(Latent Generative Landscapes):潜在生成景观,构建这个模型的目的是对之前研究的补充,之前的研究只针对真实序列数据在潜在空间的分布,但并没有过研究潜在空间本身的形状;另一方面作者希望把LGL作为一种研究框架,对蛋白质的功能,演化,多样性进行探索.
3. Results
3.1 LGL captures phylogenetic, functions, and fitness information
接下来我们来看一下作者提出的这个潜在生成景观是怎么定义的,以及如何计算的.
潜在生成景观是这样一个三维空间,其中的两个维度是VAE的潜在空间的两个维度,这两个维度所确定的平面构成了序列空间,其上的一个点可以用于表示一个特定的序列;第三个维度是代表适合度的哈密顿能量,我们可以把它看成是一个适合度地形图.
构建这样一个空间有3步,首先用蛋白质家族的多序列比对作为训练数据训练VAE,其次用同样的蛋白质家族的多序列比对作为训练数据训练Potts model.
最后我们在潜在空间构建一个网格,举个例子我们选取了一个方形区域占据了潜在空间7*7的面积,囊括了我们所有训练数据占据的空间,我们选取一个刻度将方形区域进行划分,形成一个500*500的网格,我们从每个小方格里随机的抽取一个点,然后将其放入解码器中,我们可以得到每个位点上氨基酸的概率分布,我们选取每个位点上以最大概率出现的氨基酸所构成的序列成为最大似然序列,这时就完成了潜在空间到高维空间的映射,第三部就是通过potts model给这些网格赋予哈密顿能量来表征网格所代表的序列的"适合度"
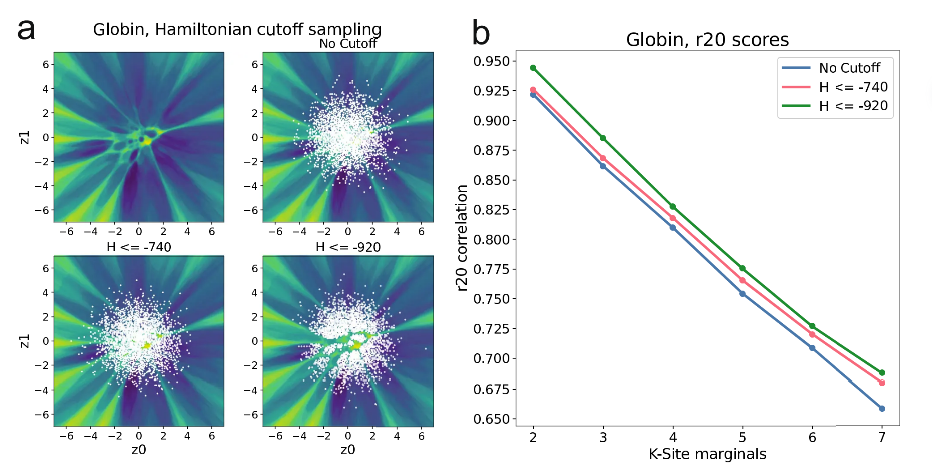
于是就有了这张图,这是一张使用珠蛋白家族的序列训练VAE得到的潜在空间,它的潜在空间的哈密顿能量的高低通过颜色的深浅来表示.我们能够观察到低哈密顿能量的区域被高哈密顿能量的序列所包围形成盆地,被用于训练VAE的序列们分布在这些盆地中,并被"山脉"分割成不同的部分.作者认为或许可以利用这个流形来改进无监督的蛋白质聚类,也就是利用模型所创造的这个空间的特征而不是仅靠数据在lantent space的坐标,比如可以设计测地线距离来衡量序列之间的远近而不是先验的采用欧式距离.
作者对这个景观做了一个初步的调查,
首先,作者希望观察最大似然序列的哈密顿能量和这个最大似然序列的ELBO是什么关系
ELBO是用来描述序列重建水平的损失函数,如果单单看一个序列它的表达式是
这是一个积分,可以用抽样的方式计算均值代替,抽样是在训练的时候进行的,但我不太认为在这个评估中作者直接这么去算,我翻了一下它的源代码,目前没有找到跟这个试验相关的脚本,所以我估计这个所谓的ELBO值(我用\(\mathrm{ELBO^*}\)和真正的\(\mathrm{ELBO}\)进行区分),它应该是这么算的
\(x_{\mathcal{L}_{max}}\)是最大似然序列,z就是作者在潜在空间网格的一个方格中选取用于代表该方格的点,这个式子相当于节省了积分的过程,不然这个计算消耗的时间和资源会很恐怖.
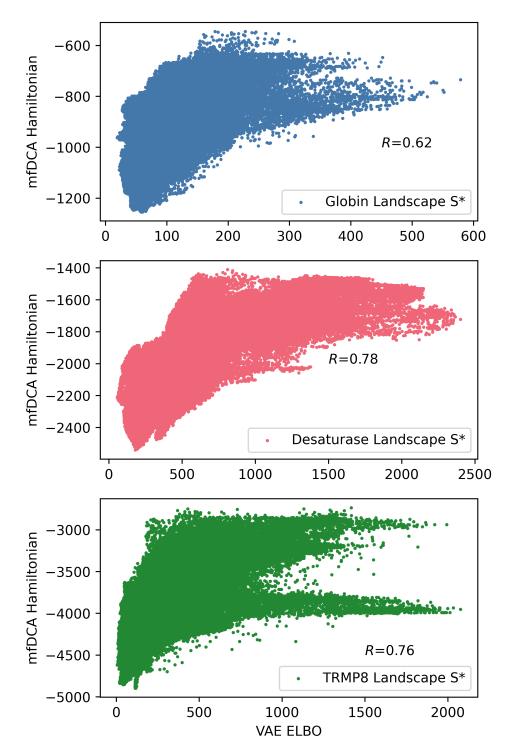
(这张图展示了作者使用的三个蛋白质家族各自所有网格内抽取的点的分布情况,横坐标是\(\mathrm{ELBO}^{*}\),纵坐标是哈密顿能量)
作者发现最大似然序列的哈密顿能量和最大似然序列的\(\mathrm{ELBO}^{*}\)是呈现正相关的,作者认为\(\mathrm{ELBO^{*}}\)也是可以作为适合度的一种表示,只不过哈密顿的生物学意义在之前的研究中已经被多次使用了,因此作者在本文中保持使用哈密顿能量.这个探索是有意义的这意味着我们有可能摆脱potts model而是仅使用VAE模型本身来构建流形.
第二个方面作者将真实序列的哈密顿能量与真实序列对应的最大似然序列的哈密顿能量进行的比较,作者在文中作者没有表明这样做的目的,愚以为作者是希望验证最大似然序列确实可以在一定程度上代表真实序列,从哈密顿能量水平上最大似然序列和真实序列不会相差太远.
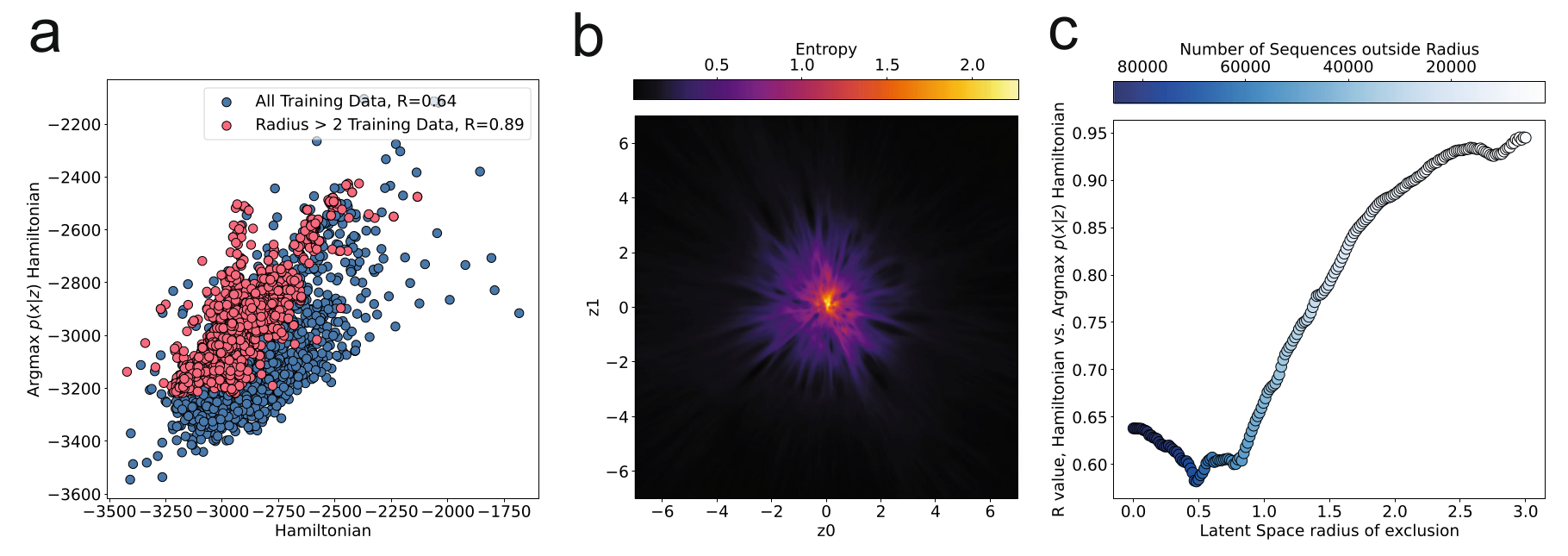
作者发现两者是呈现正相关的,从珠蛋白家族来看这个系数可以达到0.64,而当作者排除那些接近潜在空间中心位置附近半径为2以内的序列后,这个系数可以达到0.89,作者发现这种相关性的提高与序列远离潜在空间的高熵区域似乎是有一定关系的.
潜在空间上一个点的熵\(\mathrm{H(seq)}\)表示为该点通过解码器解析为序列每个位点上氨基酸的概率分布后,每个位点上的多项分布的香农熵的总和
\(N\)为序列长度,\(p_{ij}\)表示的是第\(i\)个位点上出现氨基酸\(j\)的概率
作者还解释了为什么使用2维的latent space,作者使用r20这样一种指标来评估latent space维度这件事,该指标可以说明一种生成模型对序列的高阶上位性的保留程度,作者发现提高latent space的维度在位点上位性的保存能力上其实没有太大的改进,也就不必用更高的维度了,另一个原因可能就是为了可视化

(四个子图图表示四个蛋白质家族,横轴表示的上位性的阶数,纵轴表示r20,绿线表示真实序列的训练集和真实序列中抽取的一个测试集相比上位性的保留水平,把它作为基线.剩下的两条线表示的是测试序列和我们在潜在空间上基于标准多元正态分布抽取的最大似然序列对应的r20)
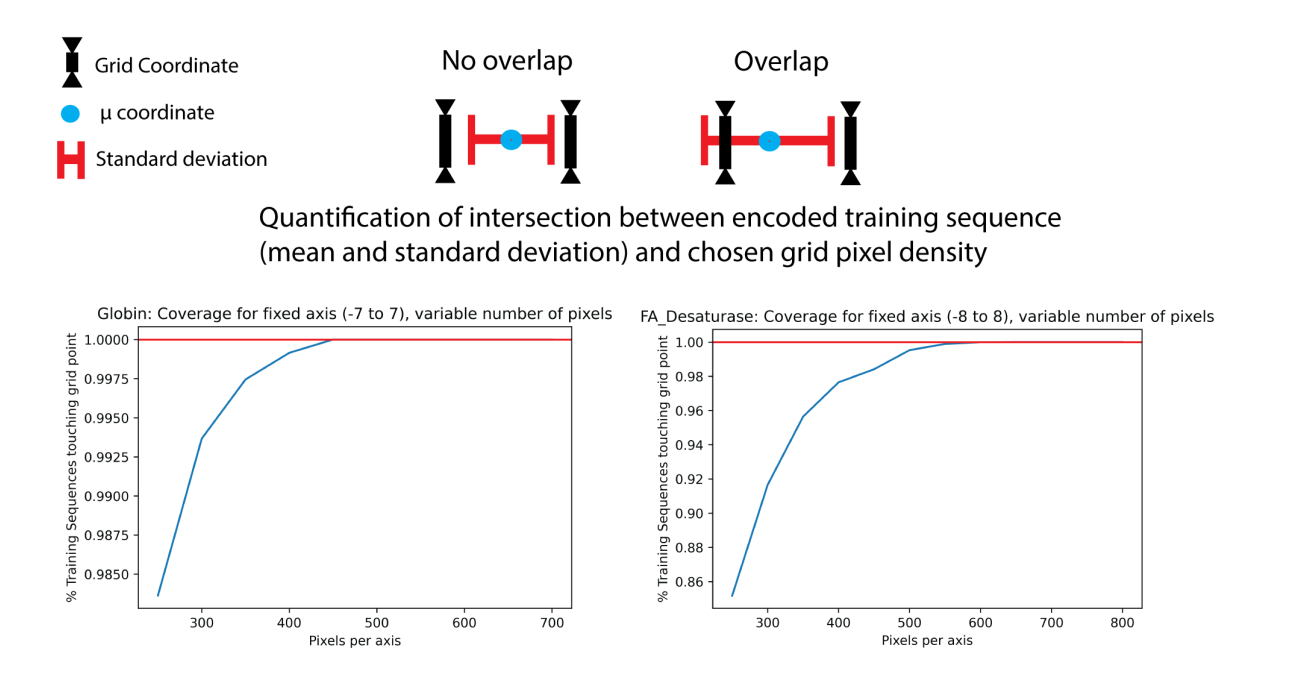
作者还解释了他是如何确定网格的刻度,总的原则就是网格的刻度要精细到,训练数据的每条序列经encoder所确定的后验分布的标准差所划定的圆周要与网格接触,作者并没有说明确立这个原则的原因
总的来说,作者经过初步的调查表明,这样一个2维潜在生成景观能够足够反映蛋白质家族序列空间,蛋白质家族特性了.你可能还注意到这张图上还标注了流线,它是怎么来的呢?作者通过将网格上选取的潜在空间的点们Z对应的最大似然序列重新输入编码器重新生成了新的潜在空间的点Z‘,于是通过Z和Z’可以计算一个梯度,通过matplotlib将其转化为流线图.通过这张图作者暗示似乎可以用这个做些东西比如演化路径,但暂时没有细说.
3.2 LGL identifies latent space for specific funtions and family-likeness
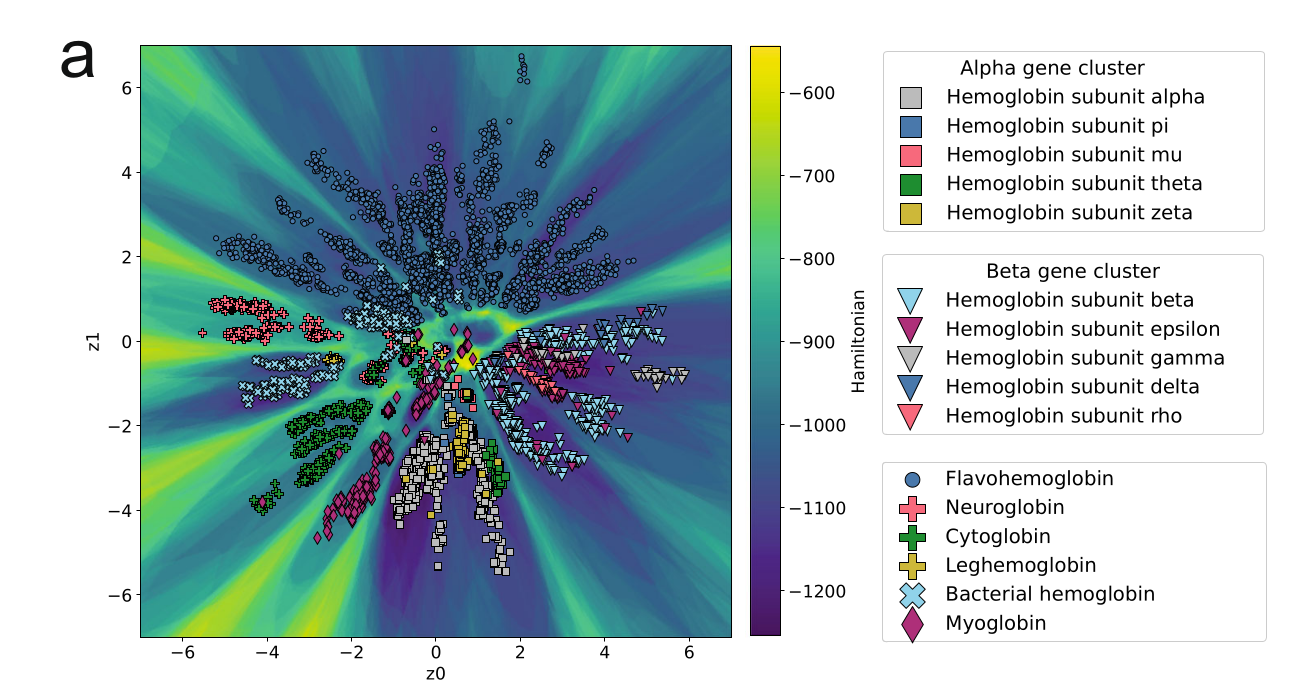
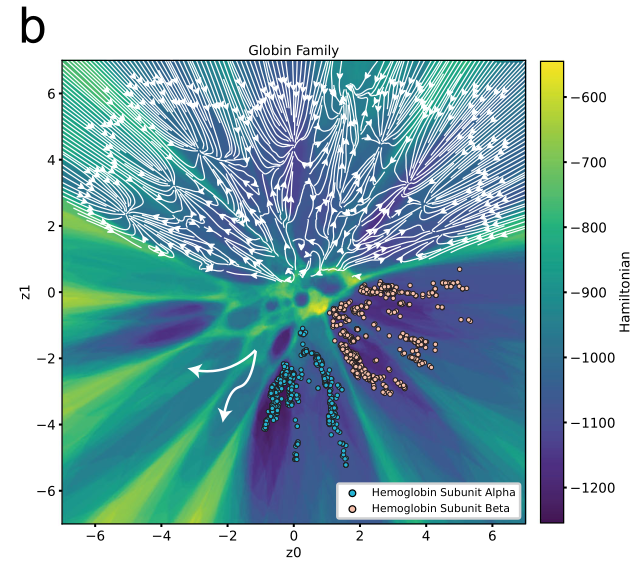
接下来作者去看了他构建的这个LGL能不能反映序列间的系统发育关系,作者选择珠蛋白家族作为对象计算了这个家族的LGL,可以观察到\(\alpha\)-珠蛋白和\(\beta\)-珠蛋白在LGL这张图上可以看到它们被划分到了不同的区域,并且在两种蛋白所在的区域之间具有一个还算清晰的能障将两个集合分隔开来,一是反映了LGL较好的分类能力也体现了\(\alpha\)珠蛋白和\(\beta\)珠蛋白内在的不同,它们在脊椎动物生长发育中具有不同的调控机制和功能.

此外在训练集中包含了很多没有清晰注释的序列,或者在分类上非常模糊。很多这些序列可以通过在一个坑里的其它序列注释,目前已经有许多方法利用vae这么做了.作者的评价是,你们这些方法需要利用输入数据先聚个类,而LGL并不需要借助这一部而是借助流行本身的特征给出一个明确的划分。此外,作者希望验证LGL这个流形保有潜在序列间的系统发育关系,作者的一个思路是验证"与真实序列对应的在流形上的最大似然序列它们所推断出的的系统发育关系和直接用真实序列推断出来的系统发育关系很像."
作者在这里使用聚类信息CI来度量生成序列和真实序列各自推断出的系统发育树之间的相似程度,如果CI的值介于0到1之间,如果CI等于0说明两个系统发育树完全一致,如果CI等于1说明两个系统发育树拓扑结构完全不像.
CI的定义可以参考文章Information theoretic generalized Robinson-Foulds metrics for comparing phylogenetic trees.

那么作者是如何进行评价的呢?以珠蛋白为例,作者使用高斯混合模型对珠蛋白的embedding进行聚类找到70个聚类中心,排除了其中6个跨度贼大的聚类中心。
作者分别从每个64个聚类所代表的正态分布中生成10个最大似然序列并进行编码,格式为cluster_id+{0-9},构成一个序列数量为640的多序列比对,使用fasttree进行系统发育重建,这棵树我们称为gen_seq_tree,抽样方法的名字我们记为gen_method
对应的作者由从64个聚类中分别随机10条真实序列,并进行编码,命名格式和gen_method一样,构成一个序列数量为640的多序列比对,使用fasttree进行系统发育重建,重建的树我们称为real_seq_tree,抽样的方法我们称为real_method
我们用一次gen_mehtod和一次real_method构成一对gen_seq_tree和real_seq_tree的实验组样本
作者生成100对实验组样本并计算它们的CI指数
作者还设立了阴性对照和阳性对照
阴性对照就是我们用一次real_method生成一个real_seq_tree,复制这棵树,将这棵树上的叶节点的排布打乱生成一个real_seq_tree_shuffled,一共100对
阳性对照就是我们用一次real_method生成一个real_seq_tree,再用一次real_method生成一个real_seq_tree'一共100对

我们可以发现实验组和阳性对照的CI值基本一致,因此作者认为这个流形保留蛋白质家族的系统发育信息
除此之外呢,作者又做了一组,作者按照生物类群对珠蛋白家族序列进行了划分,同样也按照生物类群对脱氢酶家族进行了划分,用real_method分别在珠蛋白家族和脱氢酶家族中抽取了珠蛋白real_seq_tree和脱氢酶real_seq_tree,一共100对,可以发现两者两棵树完全不像.因为刚才的结论是流形所得到的系统发育信息和真实序列所具有系统发育信息基本一致,另一方面又因为不同蛋白质家族的真实序列间按照生物类群划分抽样构建的系统发育树仍有很大差异,所以推理得到LGL学习的是蛋白质家族特异的以该基因旁系同源主导的系统发育信息.
最后作者指出流形对系统发育信息的保留体现在在盆地间高能障的存在,当我们从一个盆地翻越山峰抵达另一个盆地的时候会伴随着解码器解析出的序列概率分布的剧烈转变.一种解释就是我们的训练集在序列空间上的分布就不均匀.作者又用r20度量评估了LGL的统计保真度,r20的作用我们在之前3.1讨论latent space维度的确定时已经说过了,作者从一个标准二元正态分布中对潜在空间进行抽样,并通过设定哈密顿阈值回避高能的点,组成一个多序列比对并计算它相对于真实序列的r20.作者发现阈值设置的越低,生成序列的保真性越高,这说明在潜在空间中高哈密顿路径会破坏序列内在的位点间的联系,而其他的位置会保留这种关系.
3.3 Local LGL encodes important information for function and fitness

作者在证明完VAE具有解析蛋白质家族系统发育信息的能力后,作者又表明蛋白质在VAE潜在空间上的局部差异能够被LGL解析.作者首先分析了LGL是怎么对beta内酰胺酶家族中class A的功能进行编码的.beta-内酰胺酶是一个能够水解beta-内酰胺抗生素beta酰胺环的一种酶,可以赋予细菌对抗生素的耐药性.这个家族可以根据水解的机理进行划分.其中classA 它是内酰胺酶家族下面的一个大支,底下又可以按照内酰胺酶的底物,盘尼西林,头孢酶素,有可以通过抑制剂进行进一步的划分,不过终归是没有一个比较好的划分标准.因此作者就希望使用VAE看能不能有一个好的划分.
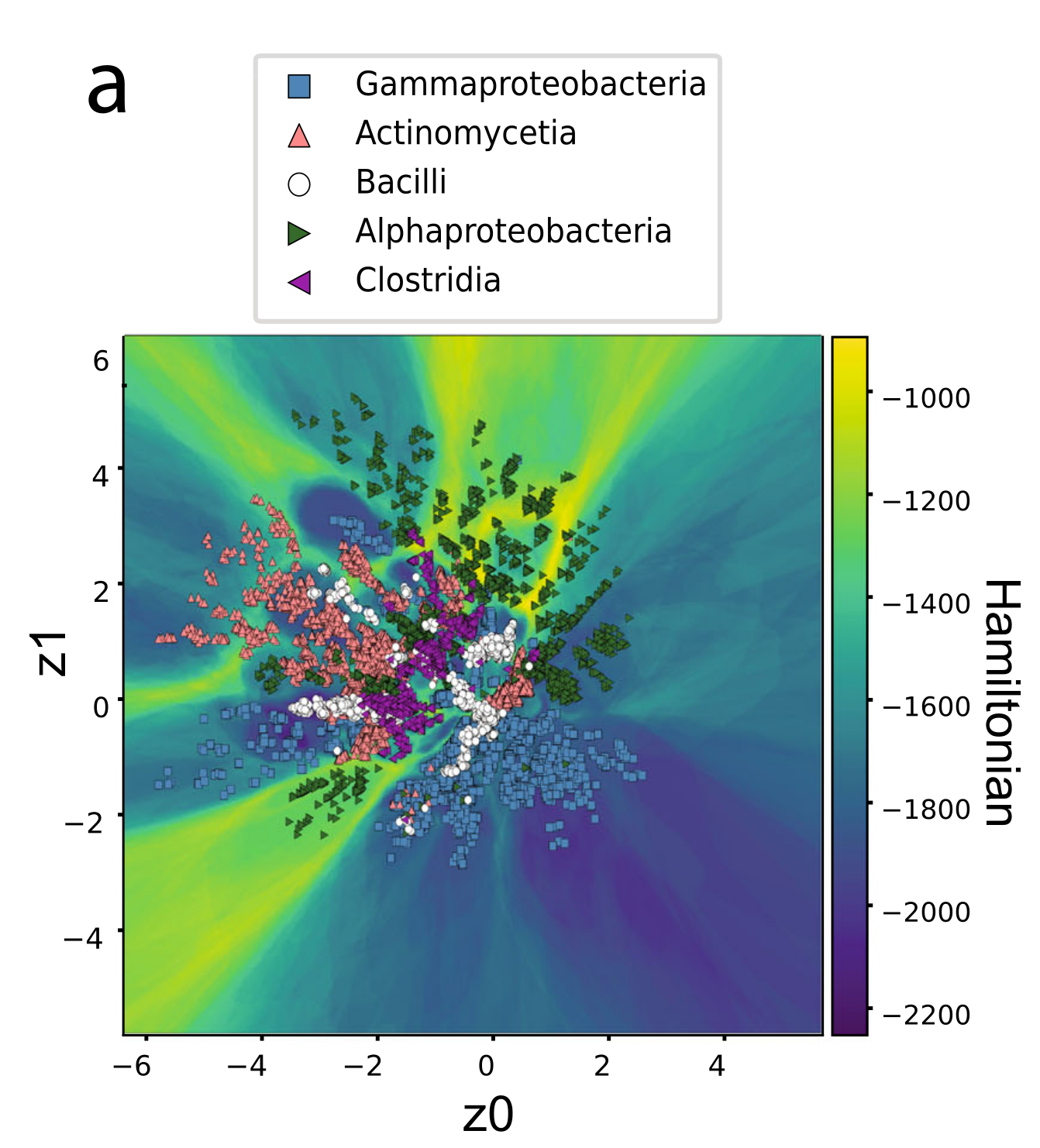
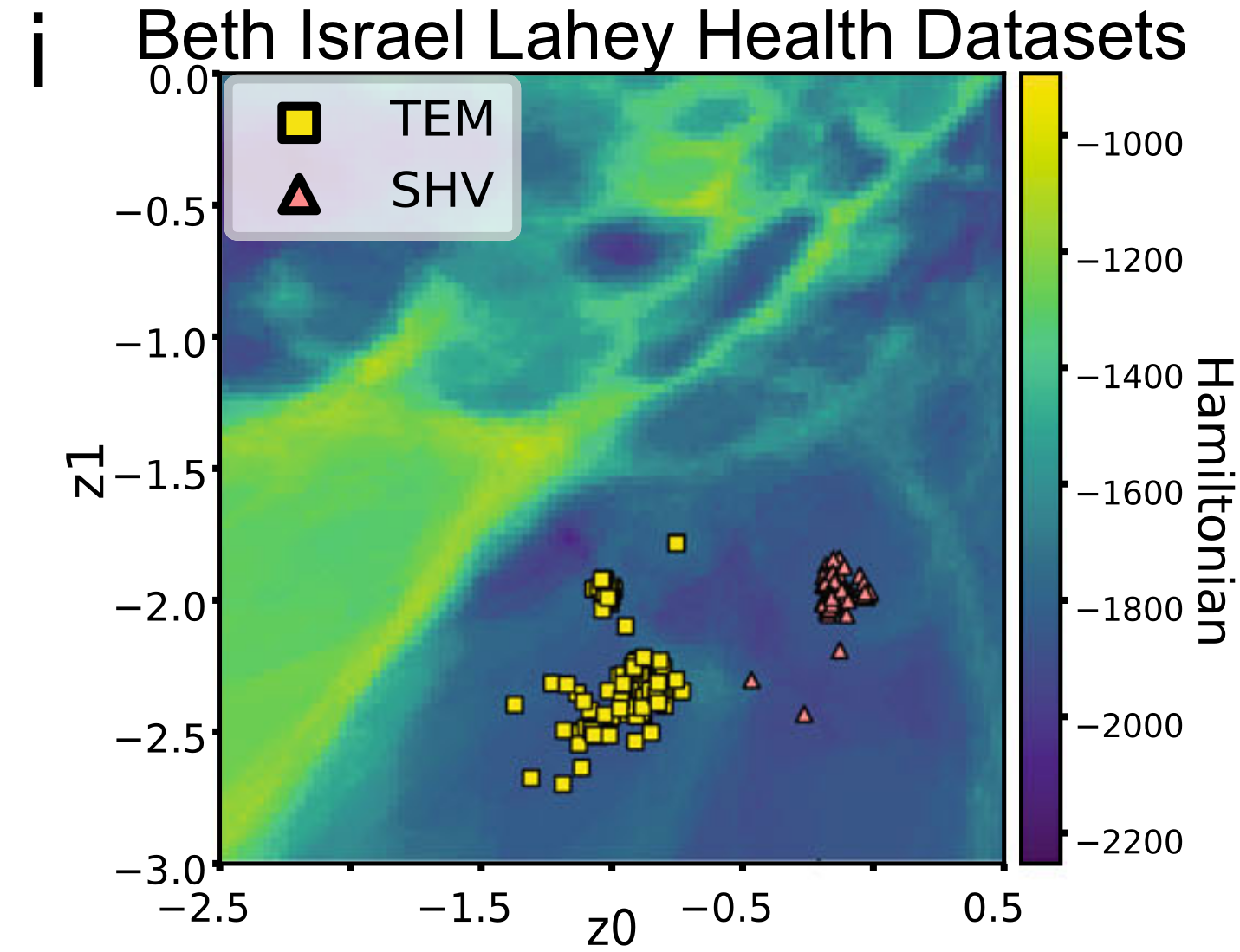
通过beta-内酰胺酶家族对VAE进行训练,并得到它的LGL(在图中作者按照生物类群对蛋白质进行了划分,gamma变形菌纲,放线菌,芽孢杆菌纲,alpha-变形菌纲,梭菌纲)在Fig 5B中可以看到TEM beta-内酰胺酶和SHV beta-内酰胺酶,这两个酶他们对广谱的头孢霉素和盘尼西林,广谱头孢霉素,单环类抗生素都具有水解能力,要想鉴别他们俩只能通过这个酶能否降解先锋霉素来进行区分.但是通过VAE呢我们发现这俩分开的较为明显了了,从LGL上看他们之间有比较微弱的能障,作者说即便这个能障不是很明显但也反映了重要的序列特征差异以及演化相关的信息。作者认为利用LGL这种分类可以对细菌的抗生素耐药性进行较为可靠的预测,此外能够预测细菌群体可能的演化的路径,有助于确定有意义的抗生素轮换方案.
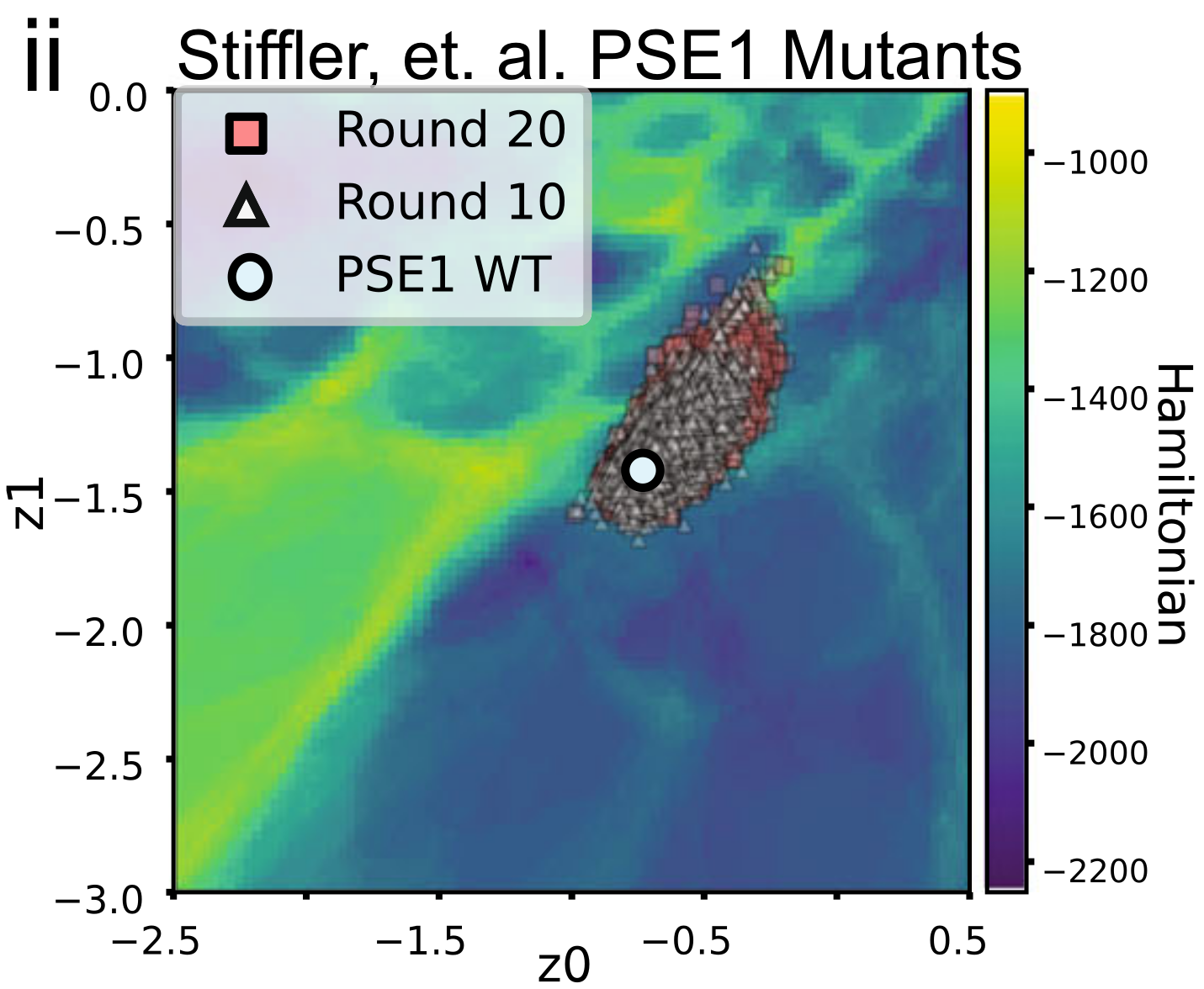

作者又进一步研究蛋白质的演化在LGL上的移动,作者使用的数据集是PSE-1(by Stiffler)和TEM-1(by Fantini)的一个实验室演化数据集,PSE-1是另一种beta-内酰胺酶它能够对羧苄青霉素的内酰胺环进行水解,TEM已经说过了,前者演化的深度比较深,与演化起始时他们的祖先平均有30%多氨基酸的差别,后者演化的深度比较浅,与起始的祖先序列平均有10%氨基酸的差别,具体怎么演化的我没有细看.作者把他们放进已经训练好的LGL去查看两个数据集的分布,可以看到TEM主要分布在一个盆地里,和野生序列的哈密顿能量差值多为负值,哈密顿能量普遍较低,而PSE1有些序列处于高能障附近,与野生型的哈密顿能量的差值为正值.关于PSE1有些序列在高能障附近,而TEM-1集中在坑里,作者说这可能是因为两个实验模拟演化的时候选择压力不同,PSE-1它使用的是6ug/ml的盘尼西林,TEM-1那一组是25ug/ml的,环境的选择压力不同,PSE-1那一组选择压力不大,也就有机会翻越能障.总之上述现象呈现了这样一种情景当环境的选择压力降低的时候,序列的突变体更容易跨越能障进入新的坑.类似的在补充材料里还有一个关于VIM-2金属beta-内酰胺酶在不同选择压力下的演化实验。总而言之,通过潜在生成景观可以反映种群的多样性和演化的趋势.

如果我们在研究蛋白质家族特定的某个基因的时候,也就是潜在景观上特定的某一个坑上进行探索的时候,作者认为可以使用LGL来观察序列之间哈密顿值的差异.跨膜蛋白8是一种离子通道蛋白(参考文献:Molecular Prerequisites for Diminished Cold Sensitivity in Ground Squirrels and Hamsters),它在很多生物体中都存在,但是仅仅在某一部分生物中对他们感受低温这个功能很重要,也就是对冷敏感.作者使用野生的对冷敏感的大鼠通道蛋白序列(简称野生冷敏大鼠序列)作为参考序列来观察冷敏感这个表型信息是如何在LGL中进行编码的.作者发现那些野生的非冷敏的松鼠通道蛋白序列(简称野生非冷敏松鼠序列)相比野生冷敏大鼠序列占据了哈密顿能量更高的潜在空间.这表明冷敏这个性状以低哈密顿的形式编码在LGL地形图中,对冷的感知能力越强哈密顿能量越低.
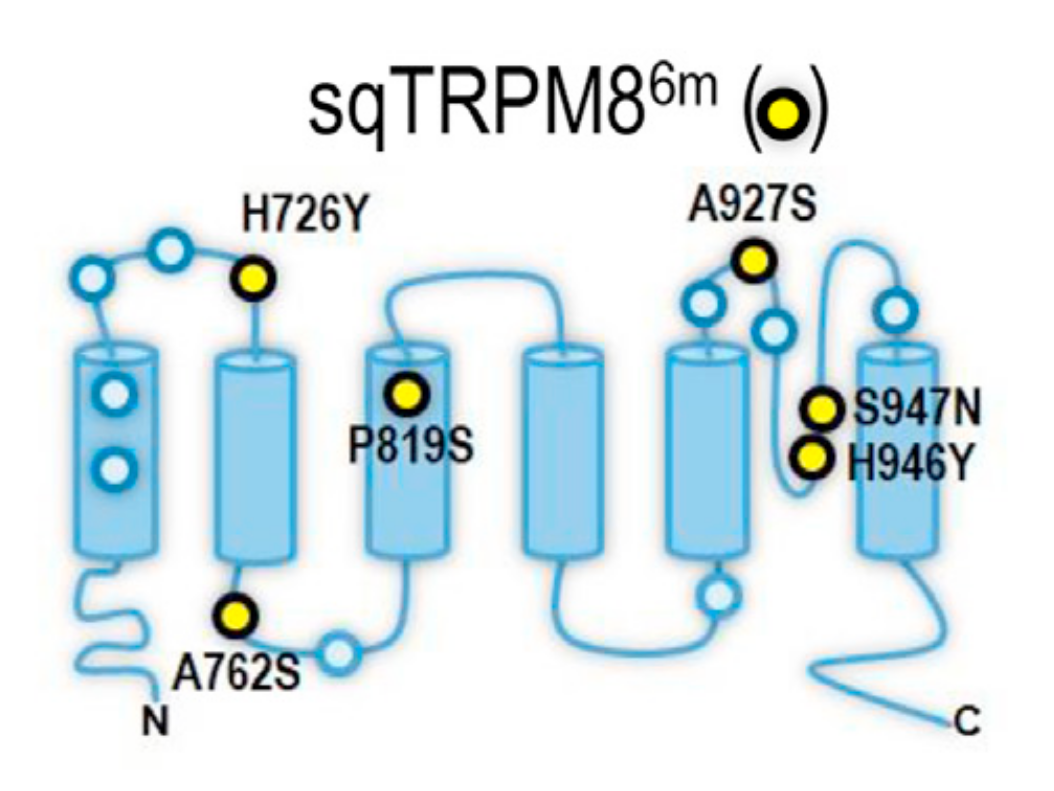

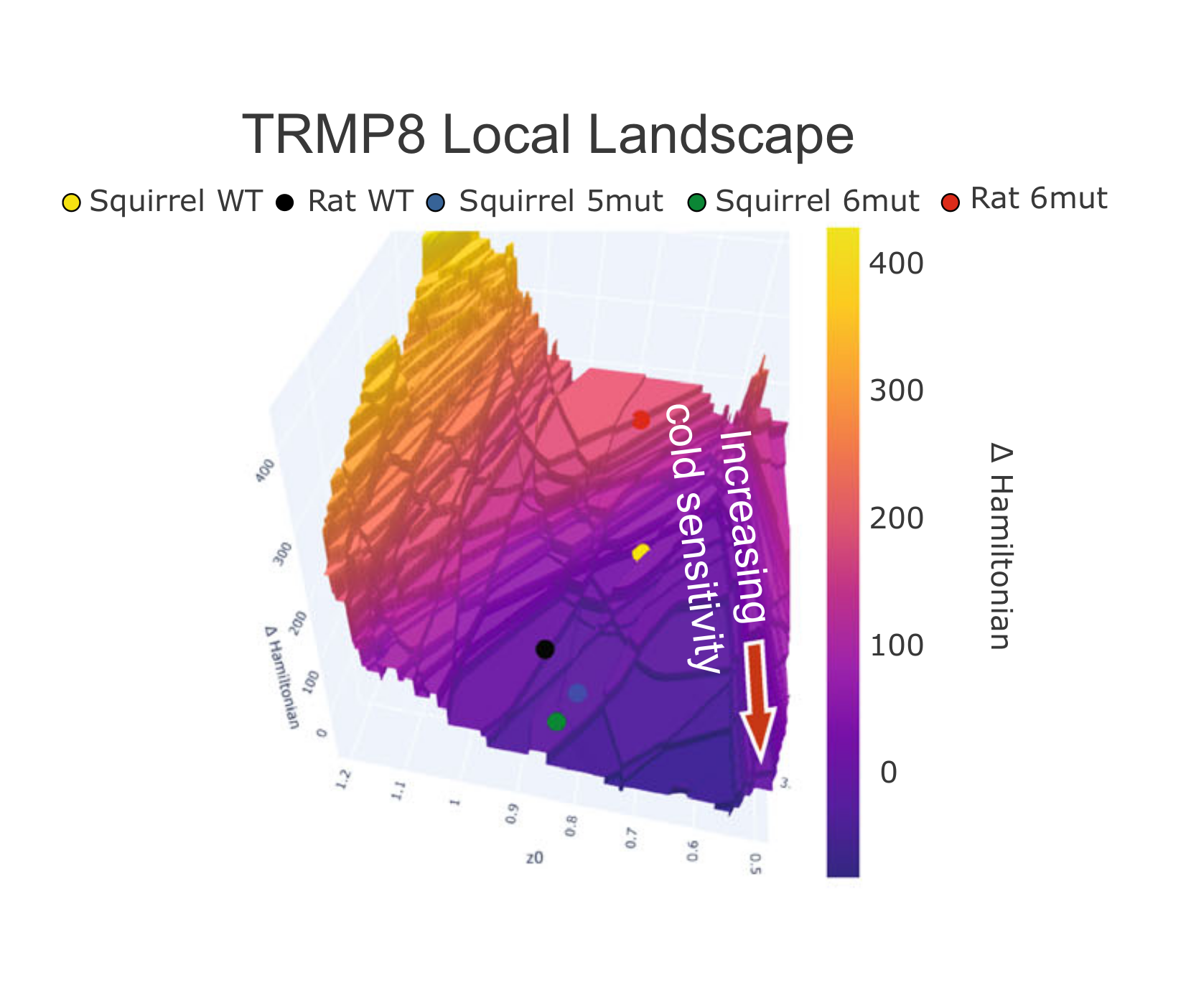
接着作者又从别的文献中得知有在跨膜蛋白8的核心区中有6个重要的位点突变对赋予该蛋白冷敏感性很重要,这是实验检验过的,怎么检验的没有具体看.然后作者将野生非冷敏松鼠上的这6个位点对应的改为野生冷敏大鼠位点上的氨基酸,输入到LGL上就发现突变蛋白一下就跑到了低哈密顿能量区。相似的,当野生冷敏大鼠序列的那6个位点对应突变为非冷敏松鼠序列6个位点的氨基酸类型的时候,突变型就直接从低哈密顿能量区跑到高哈密顿能量的位置,而且6位点突变比5突变位点在坑中的位置要更深一些,它在图中只显示了一个,而不是把五种组合都展示出来.总而言之,作者想说LGL的上的哈密顿能量分布能较好的表征通道蛋白8的冷敏特性,具有冷敏特性的通道蛋白8能够深入这个盆地,而缺乏冷敏特性的突变体会远离盆地。作者认为可以基于LGL对靶基因所处盆地进行采样来指导蛋白质工程.

.


我们现在观察到当一个序列局限在一个池子里运动的时候,我们能够对它的特性进行特性进行预测,就像通道蛋白8一样.除此之外,作者在糖苷键水解酶上又观察到局部的哈密顿能障能将序列空间分割成具有截然不同功能特征的池子.D2-beta糖苷键水解酶由于它的糖基化活性而受到人们的关注.在一篇文献(Improvements of the productivity and saccharification efficiency of the cellulolytic β-glucosidase D2-BGL in Pichia pastoris via directed evolution)中有记载,他们发现了几个比野生型D2-beta糖苷键水解酶具有更高糖基化效率的突变体.在上图中展示野生型D2-beta糖苷水解酶和它的突变体的分布.可以观察到没有突变体是正好落在能障上的.这里作者标记了经过实验测定产物转化效率最高的突变体MutM它所处的LGL中的方位,这个突变体它的表达量是野生型的2.7倍,它这么高是因为它除了增强了酶活还增强它自己蛋白质折叠的效率,减轻了内质网的压力而其它酶提高产物转化效率的方式仅仅是通过提高酶活.我们可以看到这些蛋白跨过了池子的能障,一部分蛋白跳到了另一个池子里.反映了LGL能障能反映序列截然不同的功能特性.这种差异如果放到很大的尺度上是观察不到的.
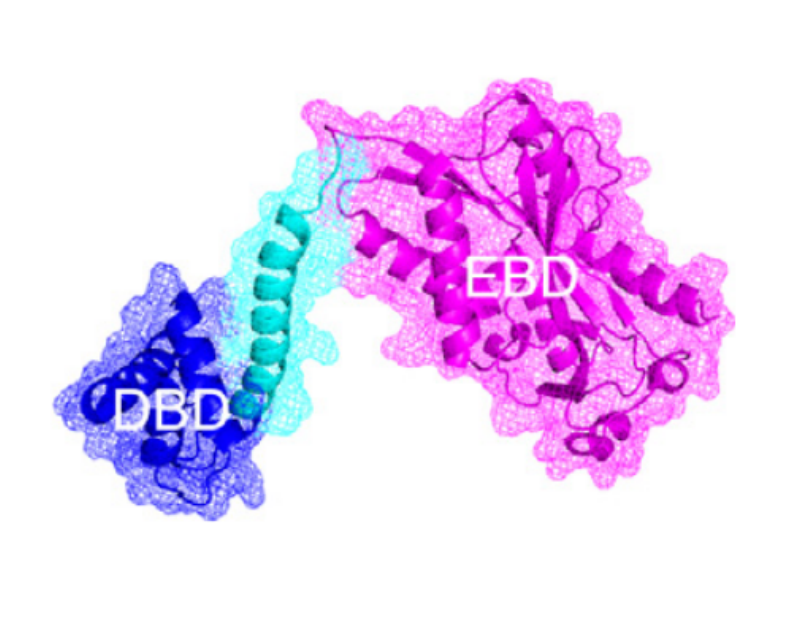

获得新的或者不同的蛋白质的特性是蛋白质工程的一个目标,但是诱变常常会伴随大量非功能突变体的产生,为了避免蛋白质重要的性质被破坏,需要交替使用正向选择和负选择。利用切换选择机制对变构转录因子进行筛选变构转录因子就是一个例子,是一类具有配体结合结构域以及dna结合域的蛋白质,当特定的小分子结合到变构转录因子上是,能够引发转录因子构象发生变化激活下游基因的表达,广泛用于生物技术,合成生物学作为生物传感器.通常需要通过大量的突变实验来筛选能够识别特定小分子的转录因子,还要保持它的对特定motif的结合活性维持稳定。在本文中涉及的这样一个变构转录因子讲座BenM(Evolution-guided engineering of small-molecule biosensors),它同时具有识别顺顺-2,4-己二烯二酸(顺顺-粘康酸)和己二酸的能力。
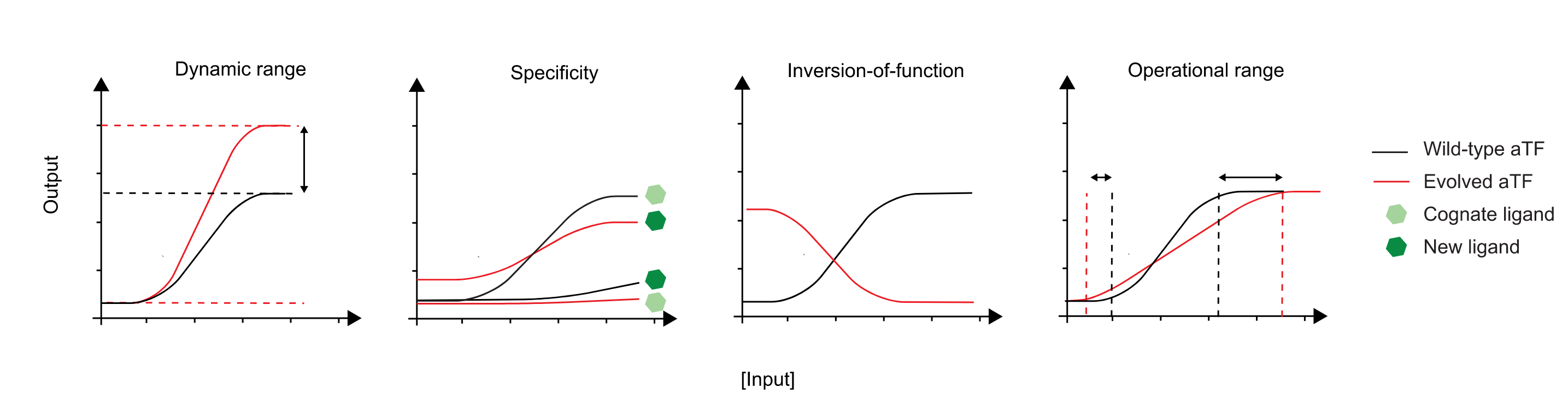
.

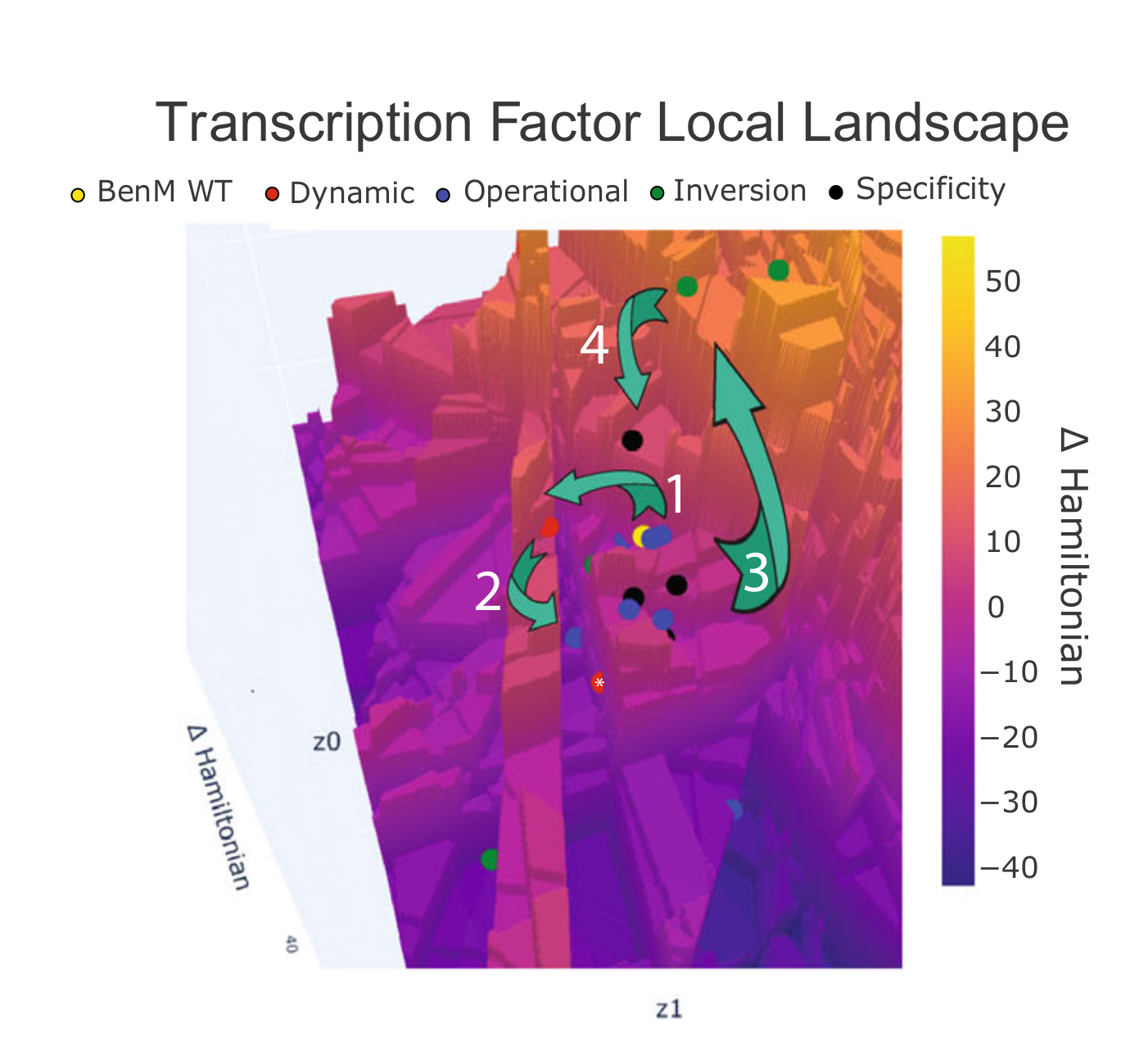
BenM的突变体库在建立过程中需要去除那些没有功能的突变体,首先建立一级突变库,那些在信号分子顺顺粘康酸诱导下使得下游基因表达量提升的突变体被称为动态突变体。这个步骤形成的突变体通过数字1在fig 6中被标注。作者使用变构蛋白质家族PF03466构建了LGL景观,在文献中使用的动态突变体的2/3位于benM盆地周围的能障上。位于盆地中的突变体MP02G10(标注星号的那个点)对下游基因表达的促进作用比野生型的benM的高出15倍,比其他的突变体都要出色.这表明对benm下游基因的稳定表达这个特征和低哈密顿能量是有正向关系的。这一轮的选择压力较低,我们看到大多数突变体占据了不太有利或中性的序列空间。
然后使用动态突变体来创建二级突变体文库,在分别用顺、顺粘康酸和己二酸处理后,在该文库中选择两种情景下表达量相近表达的突变体。这些突变体被指定为可操作范围突变体(operational mutants),这些突变体又回到了盆地里。这支持了这样一种观点,即选择压力的增加会驱使突变体向景观的低海拔移动。
我们从操作范围突变体产生三级文库,如图6f中的(3)所示,选择己二酸比顺式、顺式粘酸表现出更高表达的突变体来产生转化突变体(inversion mutants)。反转突变体经历较少的选择压力,又离开局部盆地,占据较少的有利空间。
然后使用反转突变体来产生四元突变体文库,其中当与用顺式、顺式粘酸处理的野生型BenM相比时,在用己二酸处理后选择突变体进行可比较的表达,如图6f中的(4)所示。从该最终文库中选择的突变体被指定为特异性突变体。特异性突变体再次向低海拔移动,表明控制序列重新恢复野生型的特性。
随后的几轮阳性和阴性选择在局部生成景观中发生了反应,较低的选择压力允许突变体向不太有利或中性的序列空间移动,而高的选择压力迫使突变体进入局部盆地。在较低的选择压力下占据较高的多样性空间模拟了在Stiffler等人的beta-酰胺酶中观察到的变化。上述结果反映了在环境选择压力变动的情况,序列是如何在LGL景观盆地内外移动.
3.3 Functional annotation of sequences using LGLs
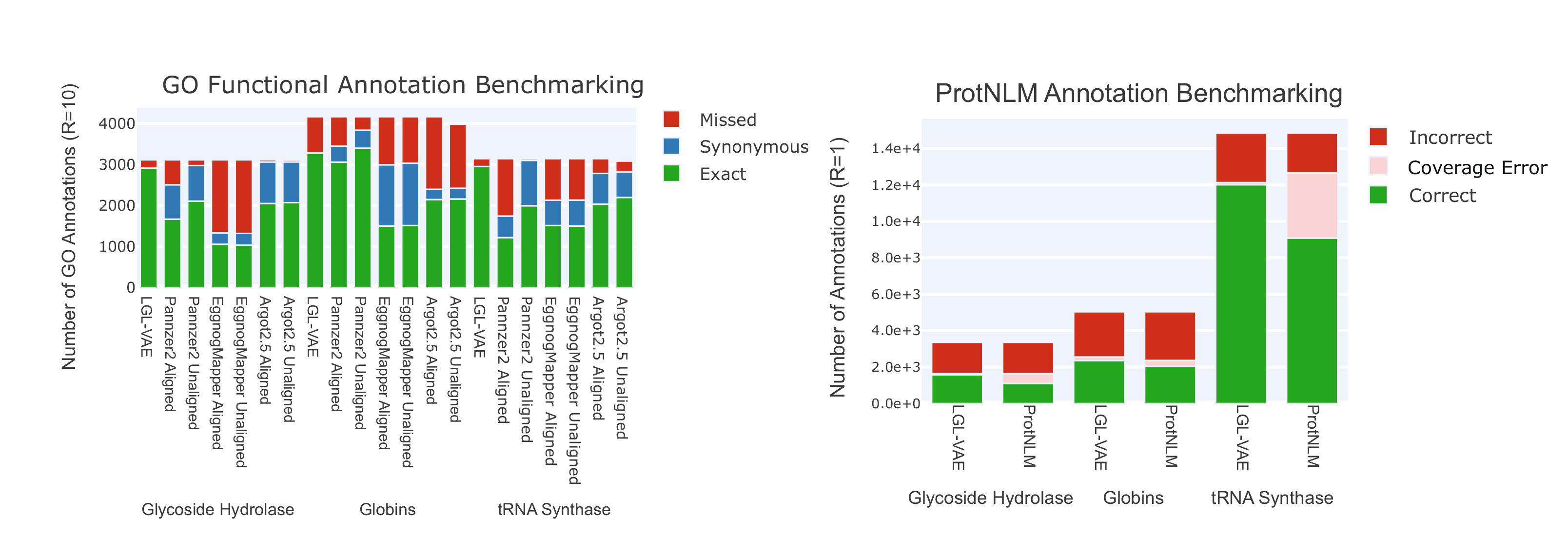
为了测试LGL在序列的功能注释中的能力,作者将LGL在功能注释上的表现与当前最先进的方法进行了比较。我在这里说一下这个注释能力是如何评估的,对于一个蛋白质家族,作者划分成训练集和测试集,训练集用于训练vae,然后将测试集嵌入到lgl中,对于每个序列找到它最临近的在训练集中的序列,用最近邻的训练集的数据对这个测试集的序列进行注释。其它的方法就是直接把测试集输入,然后给出序列对应的go注释。我们把预测的注释和测试集原有的注释进行对比,看测试集上做对的比例。作者分别做了两个实验,一个是将它和一些基因注释工具进行了比较,第二个实验是将LGL和最近开发的大型语言模型ProtNLM进行比较。如图7a所示,蓝色表示一种方法发现的注释和标签的注释同义,就是意思相同,可以看到变分自编码器的注释能力还是不错的.除此之外为了避免输入序列对性能的影响,Argot2.5、Pannzer2和eggNOG-mapper都使用了去掉gap和没去除gap的输入序列,这仅对Pannzer2产生了明显影响。在比较LGL和ProtNLM的语言模型注释时,LGL在分析的蛋白质家族中表现稍更好。这个coverage error是指protNLM产生的标签在训练集的所有注释中都没有出现且没有意思相同的.incorrect就是标签在训练集的所有注释里但是标错了.这些发现表明,潜在的生成性景观也可以可靠地用于序列的功能注释。
3.4 The latent landscape as an evolutionary map
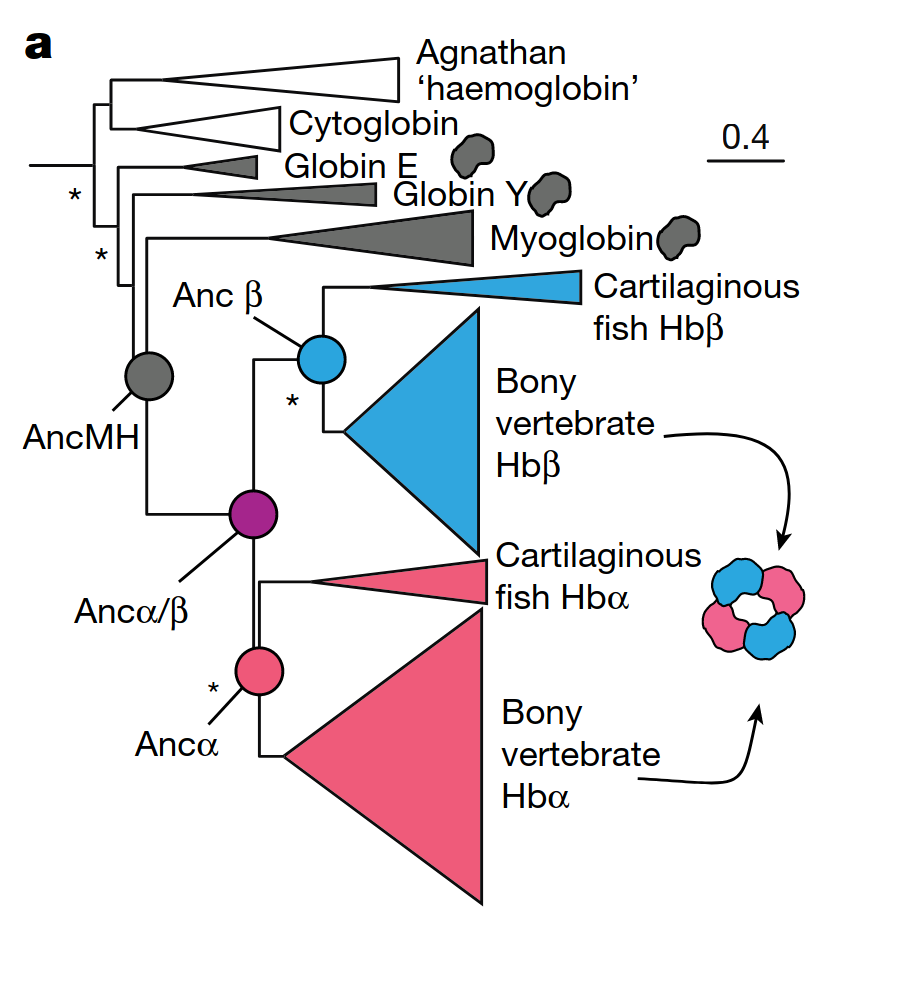

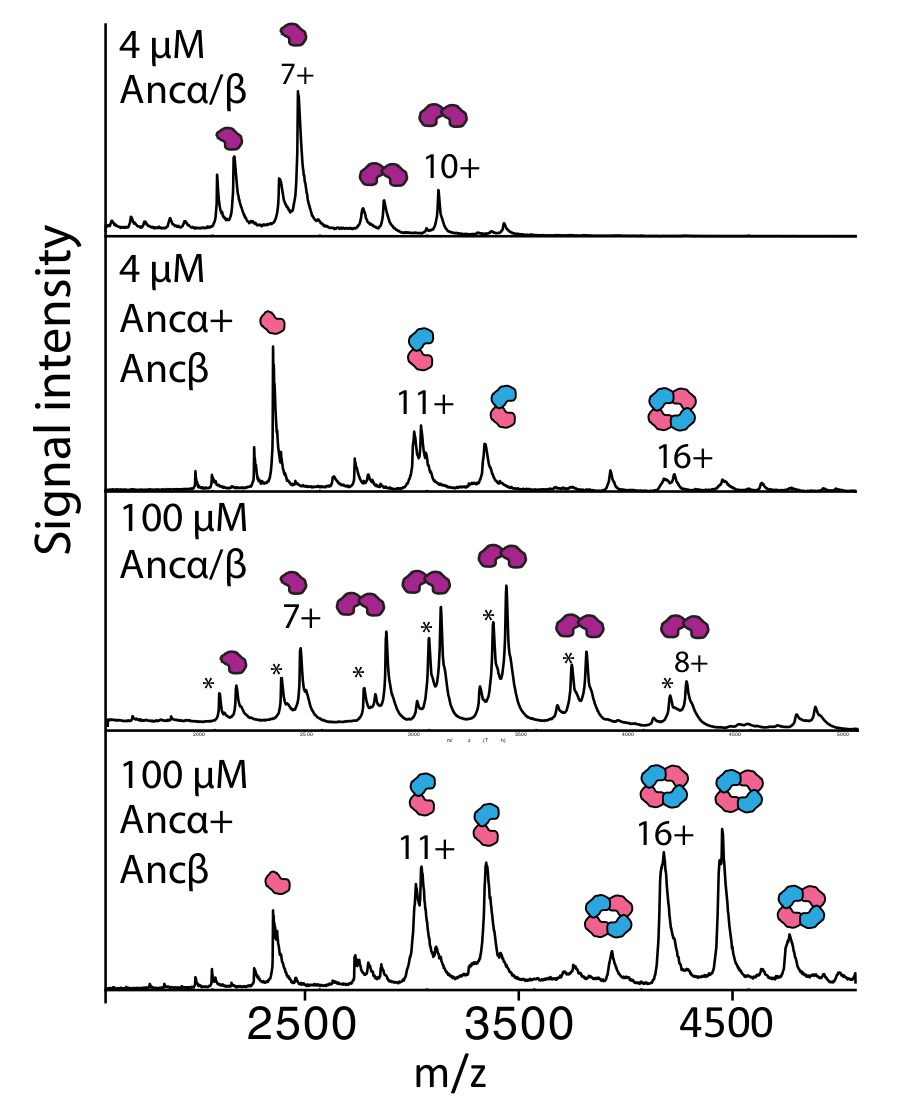
除了蛋白质家族的功能划分和适合度预测,lgl也能够一种工具来研究蛋白质的演化路径。一个例子包括对祖先血红蛋白序列进行工程设计,以获得α和β血红蛋白序列变体的功能性异四聚体.我简单交代一下背景,有一篇文献((Origin of complexity in haemoglobin evolution)[https://doi.org/10.1038/s41586-020-2292-y]),该文献的作者通过paml最大似然法重建了血红蛋白亚基alpha和血红蛋白亚基beta各自的共同祖先,Anc-alpha和Anc-beta,以及这两个共同祖先的共同祖先Anc-alpha/beta,也就是alpha蛋白和beta蛋白基因复制前夜的状态还有肌红蛋白和血红蛋白的共同祖先,探索血红蛋白异源四聚化的遗传机制和结构机制.anc-alpha 和anc-beta就已经能完成多聚化了,anc-alpha能同源二聚化,anc-beta能发生同源四聚化,这俩祖先放在一起可以异源四聚化,而基因复制前夜的这个祖先只能二聚化也就是和alpha蛋白很接近,再往前血红蛋白和肌红蛋白的共同祖先不能发生多聚化,以单体的形式存在。
alpha和beta的祖先蛋白和祖先beta蛋白一共有41个突变,有14个重要的氨基酸替换(我并不知道它比的是哪个物种的beta蛋白),完成这14个氨基酸替换就可以四聚化了.在经历了特定的9个突变后此时蛋白质叫做anc-alpha/beta 9,它可以形成四聚体了但是不可溶.作者将这个通过连续突变转变为beta蛋白的过程嵌入到vae潜在空间中,作为对照还有一些从41个差异位点上随机选取14个替换成对应在beta蛋白上的氨基酸
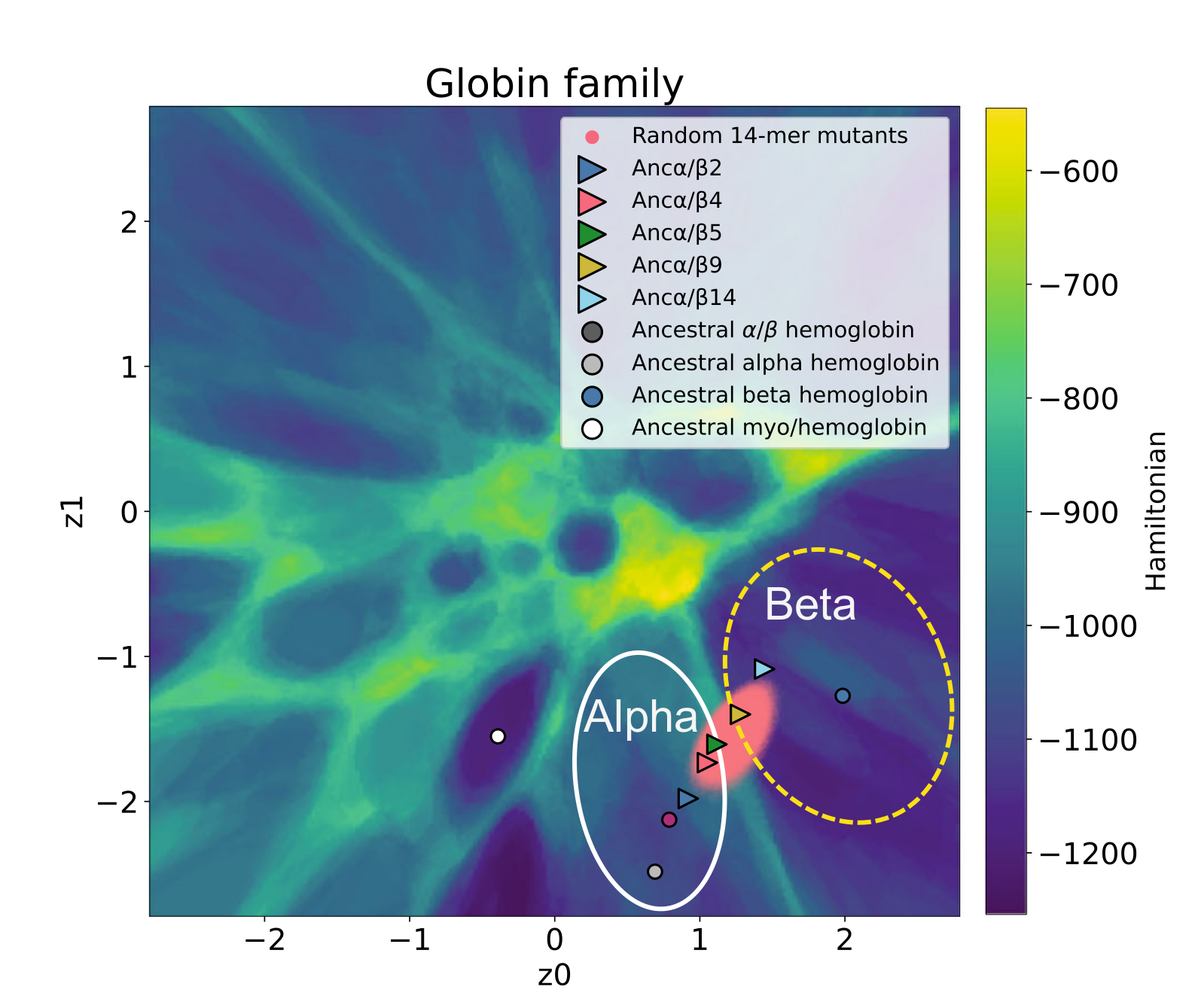
我们可以看到这样祖先蛋白从alpha蛋白盆地翻越能障抵达beta蛋白盆地这样一个过程,这与之前那篇文献描述的实验结果是相符的.随机选取位点进行突变的基本都停在能障上了。但也有一些进入beta盆地了,作者对41个位点随机选取14个的突变体中,最接近祖先beta蛋白的序列的100个突变体,和最接近祖先alpha蛋白序列的前100个的突变体他们发生替换的位点分布情况进行了统计,发现依靠随机突变且非常接近祖先beta蛋白的突变体,他们发生替换的位点和之前实验验证过的14个位点在一定程度上是吻合的,说明还有一些潜在的演化路径能完成这种转换。

作者还认为认为这些能障的高度可以为序列演化的距离提供一些量化的信息.作者以冠状病毒家族作为研究对象,把冠状病毒家族刺突蛋白放到潜在空间中呢,我们可以看到这个能障的分布和家族成员之间的系统发育关系基本一致,就是说这些能障把不同的冠状病毒给隔离开了. 从lgl图上可以看到从中东呼吸综合征到新冠病毒相比非典到新冠病毒需要经历一个高能障,也就是要打破原有的很多的序列内上位性才能达成,这反映三个beta病毒之间的演化距离,SARS-CoV2簇包含来自NCBI SARS-CoV2存储库的所有非冗余序列,并且它们围绕早期的SARS-CoV2序列呈放射状分布.该图谱还显示,蝙蝠冠状病毒序列(白色箭头)与大流行期间测序的大量变体之间不存在明显的障碍,这与蝙蝠可能是人类和动物 SARS-Cov2 之间的联系的观点一致.
Bat BCV(蝙蝠牛冠状病毒),Pangolin BCV(穿山甲牛冠状病毒),human SARS-Cov(人非典冠状病毒),Civet SARS-Cov(果子狸非典冠状病毒),Human MERS(人中东呼吸综合征冠状病毒),Bat MERS(蝙蝠中东呼吸综合征冠状病毒),Hedgehog BCV(刺猬牛冠状病毒),Murine BCV(老鼠牛冠状病毒)
4. Summary
总结一下,作者基于potts model和VAE设计了LGL(潜在生成景观)并通过几个例子,反映了该景观在蛋白质功能,分类,适合度以及演化上的生物学意义
- generative functional landscapes diversity sequencegenerative functional landscapes diversity probability-generating-function generating-function-editor generating functions binomial theorem probability-generating-function probability generating-function-editor generating function generate function vscode tests generated functions topology by diversity diverse