基于多位点序列分型技术实现假单胞菌种水平鉴定
前言
假单胞菌属的菌株为革兰氏阴性、杆状、普遍存在的细菌,其特征是营养需求低,存在于各种环境(土壤、分解中的有机物质、大气粉尘、植被和水)中。其中荧光假单胞菌与食品腐败变质密切相关。许多研究表明,假单胞菌作为主要腐败菌属,广泛存在于需有氧储存的冷藏肉[1]、鱼类[2]、乳制品[3]、新鲜农产品[4]中。在冷冻肉、鱼和牛奶中生长的假单胞菌属具有一系列有助于它们在低温条件下生存的特性,凭借其强大的蛋白水解和脂质分解解活性,使它们能够更有效地利用食物中的营养物质[5]。因此,建立一种快速准确鉴定假单胞菌的方法十分必要。
近年来,关于假单胞菌鉴定的研究不断有被报道。常见的分子生物学方法例如,DNA-DNA杂交[6],RNA-DNA杂交[7],rep-PCR(重复序列聚合酶链式反应)[8]和PFGE(脉冲电场凝胶电泳)[9] 等。目前应用于假单胞菌鉴定最常用的技术之一是16s rRNA基因测序技术,但是假单胞菌属目前已有319种被报道[10],且16s rRNA基因的分辨率通常太低而无法区分密切相关的物种,基于单基因的鉴定方法不适合假单胞菌鉴定。
自1998年以来,多位点序列分析(Multilocus Sequence Analysis,MLST)被认为是一种精确的菌种鉴定方法[11],将基因序列进行串联,增加有效信息位点数量,以便提高分辨力,对难以区分的相似菌群进行鉴定。例如,Yamamoto等人通过分析gyrB和rpoD基因来研究假单胞菌属[12],Hilario等人开发了一种基于atpD、carA、recA和16S rRNA测序的方法对食源性假单胞菌进行了分型[13]。2010年,Mulet等人介绍了一种MLST方案,通过对107株假单胞菌类型菌株的16S rRNA、gyrB、rpoB和rpoD基因测序来揭示假单胞菌属内的系统发育关系[14]。基于前人研究,本篇博文基于假单胞菌保守基因glnS、gyrB、ileS、nuoD、recA、rpoB和rpoD串联构建系统发育树,以达到假单胞菌种水平鉴定的目的。
本篇博文所采用的分析流程如图1所示

图1 假单胞菌MLST分析流程
本篇博文所使用的软件如下
Mafft(v7.520)、Trimal(v1.4)、Seqkit(2.4.0)、IQtree(2.2.2.3)、Notepad++(8.1.9.0)
具体分析步骤如下
1 数据收集
1.1 假单胞菌16S rRNA基因收集
本篇博文所使用数据均来自NCBI数据库,所收集的数据如下,388株假单胞菌16S rRNA基因,平均长度为约为1490bp,原始文件见附件1。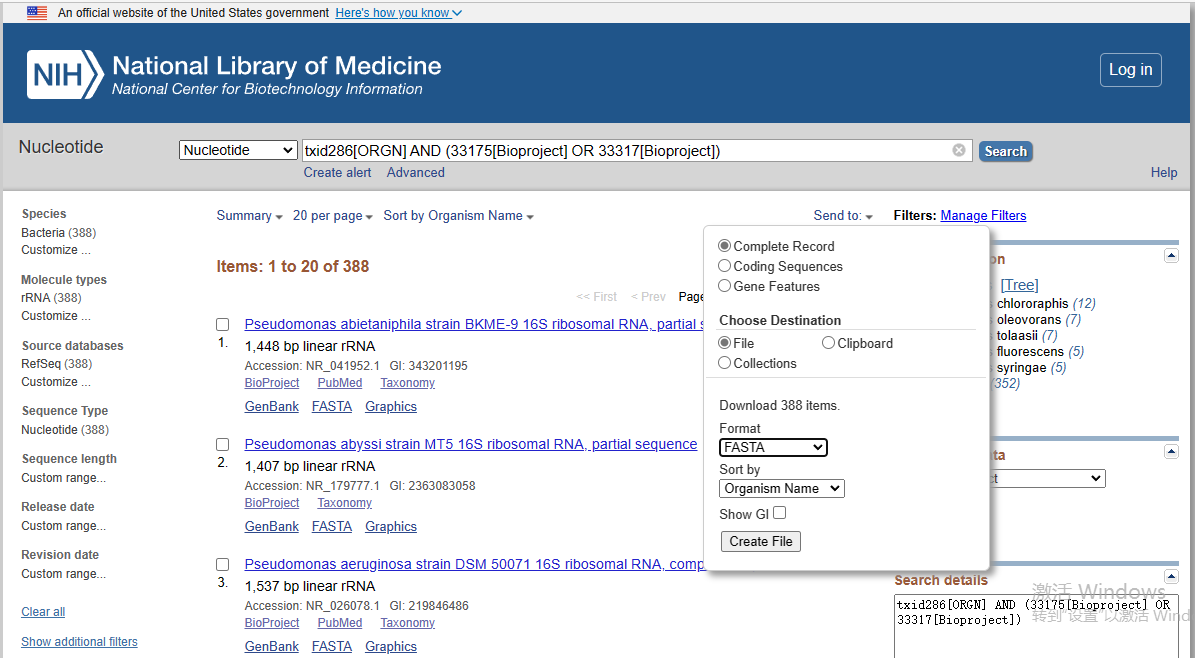
图2 16S rRNA基因原始数据收集
1.2 假单胞菌管家基因收集
18株假单胞菌属细菌的其管家基因glnS(510bp)、gyrB(492bp)、ileS(552bp)、nuoD(516bp)、recA(435bp)、rpoB(477bp)和rpoD(480bp),原始文件见附件2。
2 多位点序列分析
2.1 多序列比对
最经典和广为熟知的多的序列比对软件是 Clustalw。但是现有的多序列比对软件较多,有文献报道:比对速度(Muscle>MAFFT>ClustalW>T-Coffee),比对准确性(MAFFT>Muscle>T-Coffee>ClustalW)。因此,使用 MAFFT 软件进行多序列比对。
输入mafft -h 查看可选择的参数
图3 MAFFT软件基本参数
输入mafft --maxiterate 1000 --globalpair ../01.rawdata/glnS.fasta > glnS_M.fasta命令,进行多序列比对。
--maxiterate 1000 --globalpair 是mafft软件最准确的方法,适合小于200条,且长度小于2000氨基酸或核酸的序列。此处以glnS基因为例,其余6个管家基因序列的比对照此方法进行。
图4 MAFFT软件运行界面
输入cat glnS_M.fasta命令查看mafft比对结果,因为输入文件的格式是fasta,因此mafft输出结果也是fasta格式,如图4所示。
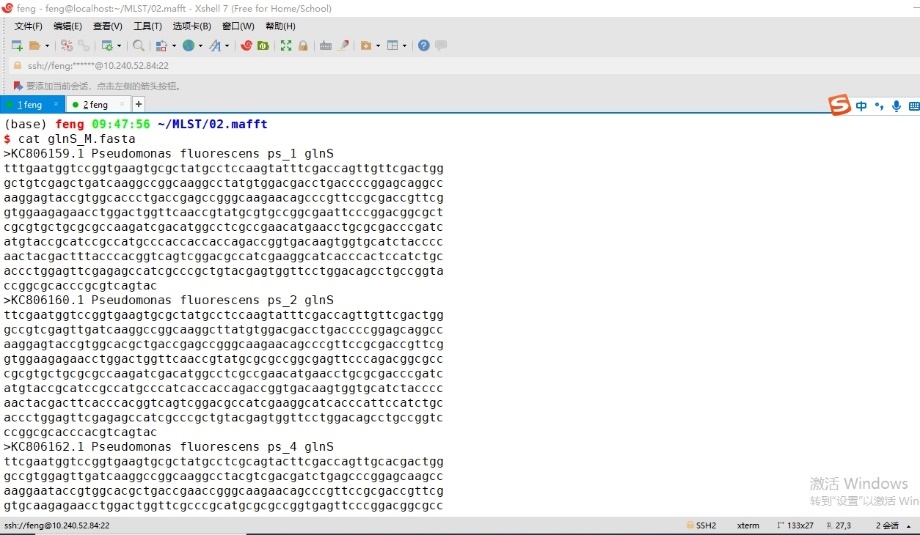
图5 MAFFT软件比对结果
2.2 Trimal软件修剪比对后的序列
使用gblocks软件修剪比对序列,可能会使大量有用的序列被裁剪掉,所以尝试使用Trimal软件进行修剪比对序列。
输入trimal -h 查看可选择的参数
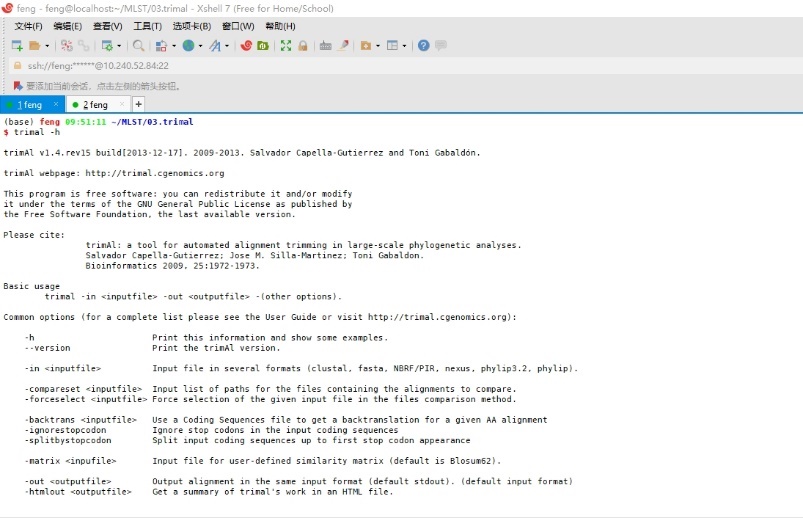
图6 Trimal软件基本参数
输入trimal -in ../02.mafft/glnS_M.fasta -out glnS_M_T.fasta -automated1命令将比对后的序列进行修剪
-in <inputfile>多种格式的输入文件(clustal,fasta,NBRF / PIR,nexus,phylip3.2,phylip)
-out <outputfile>以相同输入格式输出对齐(默认输出格式)。包括其他多种输出格式,比如:
-clustal CUSTAL格式的输出文件
-fasta FASTA格式的输出文件
-nbrf NBRF / PIR格式的输出文件
-nexus NEXUS格式的输出文件
-mega MEGA格式的输出文件
-phylip3.2 PHYLIP3.2格式的输出文件
-phylip PHYLIP / PHYLIP4格式的输出文件
此处使用mafft比对后的序列/glnS_M.fasta为例使用Trimal,由于Trimal软件运行过快且修剪序列较少,因此没有运行界面。
输入cat glnS_M_T.fasta,查看修剪后的结果。
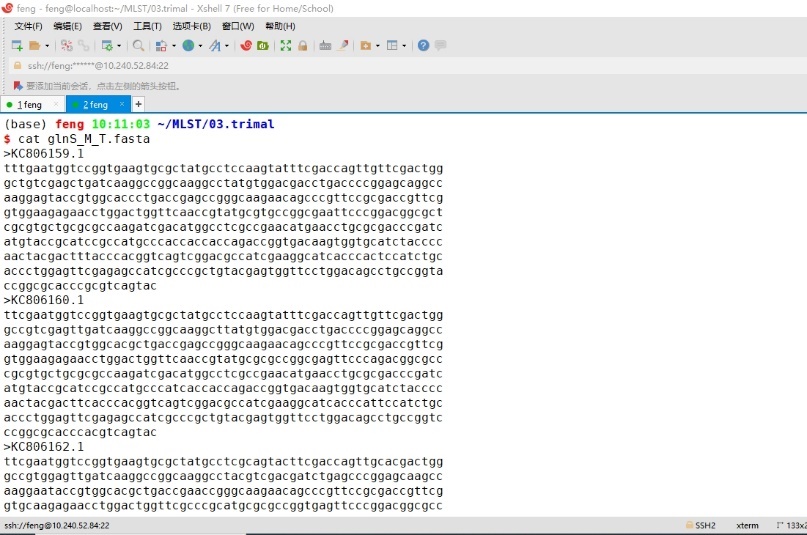
图7 Trimal软件修剪结果
2.3 Seqkit软件和paste命令联用将序列串联拼接
Seqkit是一款专门处理fsata/q序列文件的软件,功能强大,部分功能见图8。

图8 seqkit软件基本功能
输入seqkit -h查看seqkit软件基本参数。
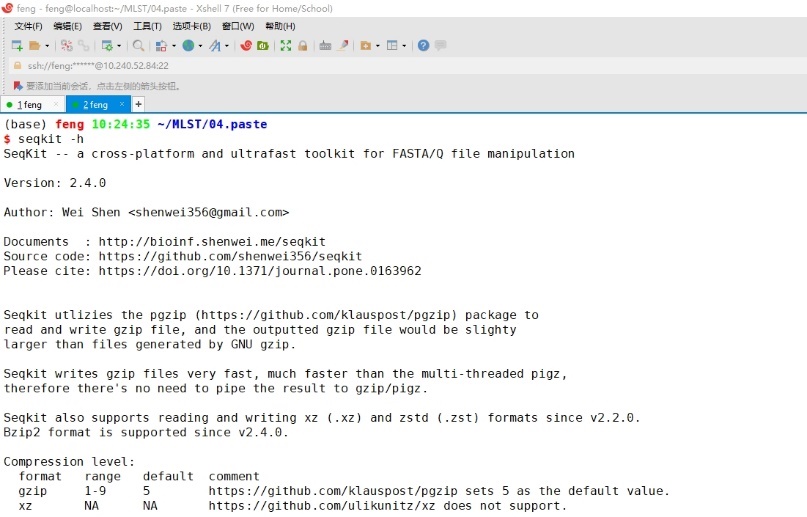
图9 seqkit软件基本参数
输入seqkit seq glnS_M_T.fasta -w 0 > glnS_M_T_W.fasta命令,将比对后的fasta序列变为单行,输入cat glnS_M_W.fasta命名,查看结果,结果见图10。
图10 将比对后的fasta序列变为单行
paste命令为linux系统自带的命令,其所使用的参数见图11。
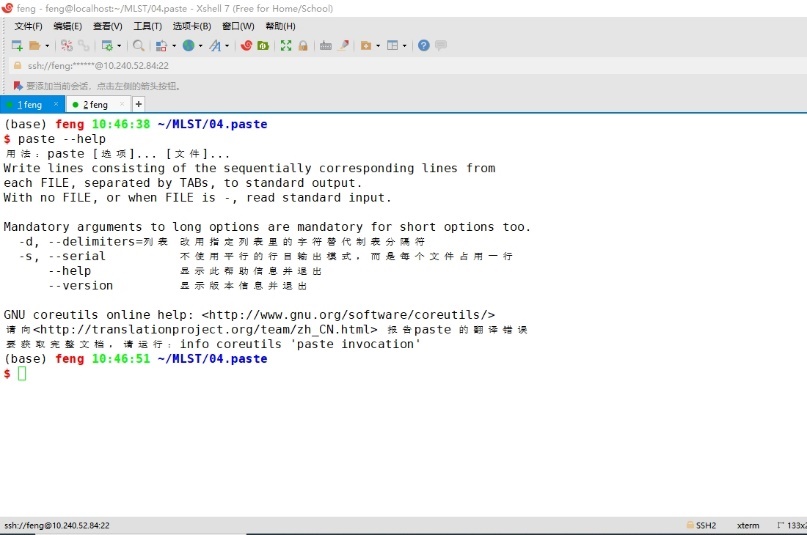
图11 paste命令基本参数
输入paste ../02.mafft/glnS_M_W.fasta ../02.mafft/gyrB_M_W.fasta ../02.mafft/ileS_M_W.fasta ../02.mafft/nuoD_M_W.fasta ../02.mafft/recA_M_W.fasta ../02.mafft/rpoB_M_W.fasta ../02.mafft/rpoD_M_W.fasta -d '' > all.fasta命令将所有比对且修剪后的序列合并。
输入cat all.fasta命令,检测合并结果,结果见图12。
图12 paste合并结果
由于fasta文件的序列头重复,因此合并后的all.fasta文件需要手动将重复的序列头删除,由于Notepad++是一款便捷强大的文本编辑器,因此使用Notepad++打开all.fasta文件,删除文件头,只保留“>pseudomonas fluorescens”文件头。
2.4 IQtree构建系统发育树
系统发育树(phylogenetic tree)构建的软件种类众多。大体上,有如下几种策略:从最简单的UPGMA法,到邻接法(neighbor joining),最大简约法(most parsimonious),再到最大似然法(Maximum likelihood),以及贝叶斯法,每种方法都有不少可以选择的工具。
IQtree是一款最大似然法发育树构建软件,输入iqtree -h查看其基本使用参数,见图13。
图13 iqtree软件基本使用参数
输入iqtree -s all.fasta -m MFP -b 1000 -nt 10 -cmax 15构建系统发育树,产生如图13所示的文件,其中all.fasta.treefile文件可提交到ITOL网站(https://itol.embl.de/)进行可视化。
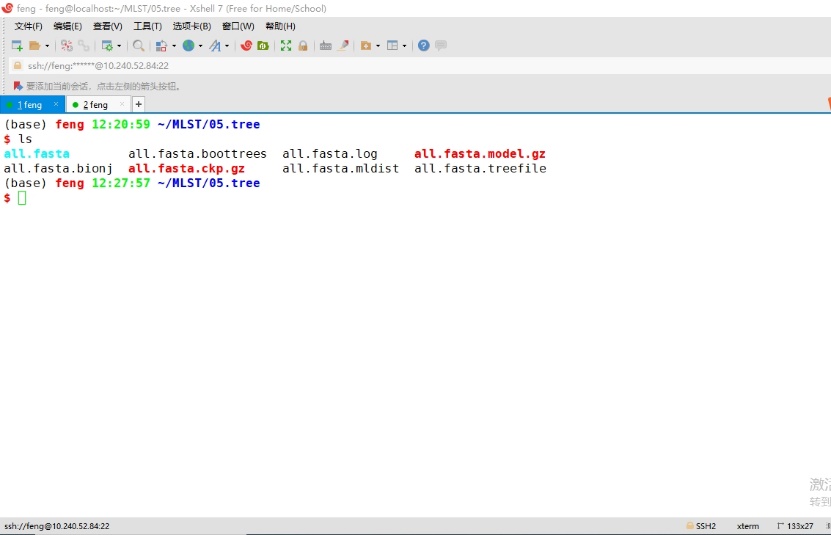
图14 iqtree软件产生的文件
2.5 ITOL可视化系统发育树
ITOL网址:https://itol.embl.de/
创建账户之后,上传all.fasta.treefile文件
图15 ITOL官网
图16 文件上传
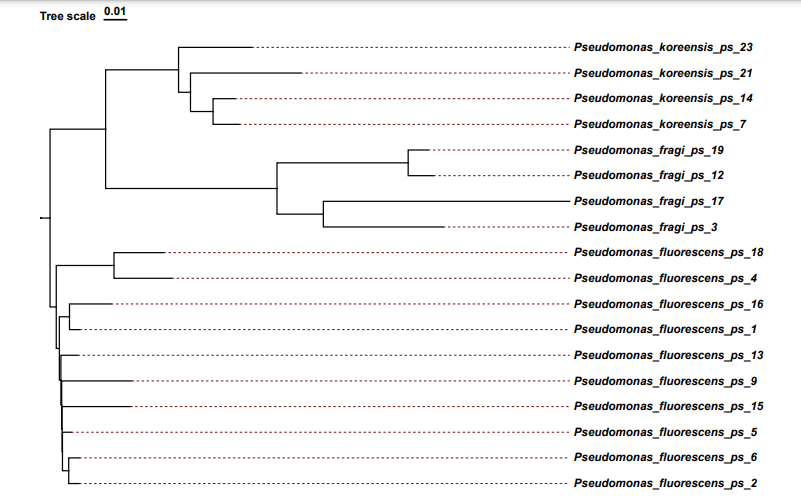
图17 系统发育树原始结构
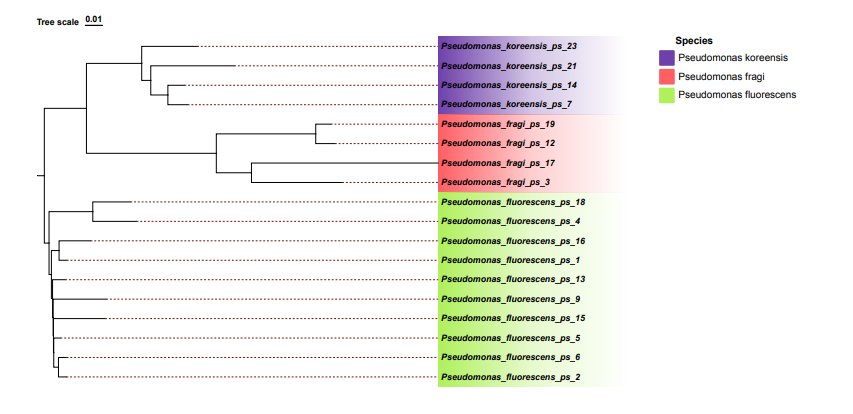
图18 系统发育树美化
如图18所示,18株假单胞单胞菌被分成三类,分别为Pseudomonas koreensis、Pseudomonas fragi和Pseudomonas fluorescens,这说明利用管家基因glnS、gyrB、ileS、nuoD、recA、rpoB和rpoD串联法构建系统发育树能够在种水平上分辨假单胞菌属内的物种。
3.基于16S rRNA基因构建系统发育树
为了更好地突出MLST的准确性,本博文将从NCBI收集得到的388株假单胞菌以及本实验室所测序的6株假单胞菌16S rRNA基因进行比对并构建系统发育树,以比较常规的16S rDNA测序能否将假单胞鉴定到种水平。所采用的分析流程与MLST基本相同,仅缺少序列修剪和拼接步骤。
3.1 16S rRNA基因比对
输入mafft -auto16S.fasta > 16S_M.fasta命令,进行多序列比对。
3.2 系统发育树构建
输入iqtree -s 16S_M.fasta -m MFP -b 1000 -nt 10 -cmax 15构建系统发育树。
3.3 系统发育树可视化
上传all.fasta.treefile文件至https://itol.embl.de/,美化后的系统发育树如图所示。

图19 假单胞菌16S rRNA基因系统发育树
注:图中深红色标注的6株细菌16S rRNA基因序列为实验室细菌送上海生工公司测序所得
由上图可知,由于16S rRNA基因分辨率较低,该基因在假单胞菌种间同源性过高,仅根据16S rRNA基因无法区分假单胞菌属内的菌种,只能鉴定到属水平。
4.总结与讨论
细菌的rRNA是细胞内含量最多的RNA,约占RNA总量的80%以上,由高度保守区和可变区组成。细菌的rRNA包括5S、 16S、 23S,其中5S rRNA信息量少,不适合分析。23S rRNA尽管分子大,信息量多,但碱基突变速度较快,同样不适用于细菌鉴定。16S rDNA 是编码原核生物核糖体小亚基rRNA(16S rRNA)的DNA序列,相比之下遗传较为稳定,长度在1500bp左右,代表信息量适中,可以用于研究系统进化。但是有的菌种由于种间差异小,单独依靠16SrDNA鉴定不能鉴定到种。需要其它鉴定方法补充。
与传统分子生物学分型方法相比,MLST具有更高的分辨力,能将同种细菌分为更多的亚型。MLST操作简单,已经用于多种细菌进化研究。随着测序速度的加快和成本的降低,以及分析软件的发展,MLST逐渐成为细菌的常规分型方法。MLST已被广泛地作为菌株鉴定的常用方法,成为一种更为准确的分型系统方法。MLST目前已经成为了细菌鉴定的一种重要方法,可通过数据库与其它国家和地区的研究结果进行比对,从而更加全面的了解细菌进化的特征。
附件1:假单胞菌16S rRNA基因序列
附件2:假单胞菌管家基因序列
参考文献:
[1] WICKRAMASINGHE N N, RAVENSDALE J, COOREY R, et al. The Predominance of Psychrotrophic Pseudomonads on Aerobically Stored Chilled Red Meat [J]. Compr Rev Food Sci Food Saf, 2019, 18(5): 1622-35.
[2] STERNISA M, KLANCNIK A, SMOLE MOZINA S. Spoilage Pseudomonas biofilm with Escherichia coli protection in fish meat at 5 degrees C [J]. J Sci Food Agric, 2019, 99(10): 4635-41.
[3] ZAREI M, YOUSEFVAND A, MAKTABI S, et al. Identification, phylogenetic characterisation and proteolytic activity quantification of high biofilm-forming Pseudomonas fluorescens group bacterial strains isolated from cold raw milk [J]. International Dairy Journal, 2020, 109.
[4] FANELLI F, CAPUTO L, QUINTIERI L. Phenotypic and genomic characterization of Pseudomonas putida ITEM 17297 spoiler of fresh vegetables: Focus on biofilm and antibiotic resistance interaction [J]. Curr Res Food Sci, 2021, 4: 74-82.
[5] DUMAN M, MULET M, ALTUN S, et al. The diversity of Pseudomonas species isolated from fish farms in Turkey [J]. Aquaculture, 2021, 535.
[6] Wayne, L.G., Brenner, D.J., Colwell, R.R., Grimont, P.A.D., Kandler, O., Krichevsky, M.I.,Moore, L.H., Moore, W.E.C., Murray, R.G.E., Stackebrandt, E., Starr, M.P.,
Trüper, H.G., 1987. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 37, 463-464
[7] Palleroni, N.J., Kunisawa, R., Contopoulou, R., Dourdoroff, M., 1973. Nucleic acid homologies in the genus Pseudomonas. Int. J. Syst. Evol. Bacteriol. 23, 333-339
[8] Johnsen, K., Andersen, S., Jacobsen, C.S., 1996. Phenotypic and genotypic characterization of phenanthrene-degrading fluorescent Pseudomonas biovars. Appl.Environ. Microbiol. 62, 3818-3825.
[9] Nogarol, C., Acutis, P.L., Bianchi, D.M., Maurella, C., Peletto, S., Gallina, S., Adriano, D.,Zuccon, F., Borrello, S., Caramelli, M., Decastelli, L., 2013. Molecular characterization of Pseudomonas fluorescens isolates involved in the Italian “bluemozzarella” event. J Food Prot. 76, 500-504.
[10] BEKHIT A E-D A, HOLMAN B W B, GITERU S G, et al. Total volatile basic nitrogen (TVB-N) and its role in meat spoilage: A review [J]. Trends in Food Science & Technology, 2021, 109: 280-302.
[11] Maiden, M.J.C., 2006. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol.60, 561-588.
[12] Yamamoto, S., Kasai, H., Arnold, D.L., Jackson, R.W., Vivian, A., Harayama, S., 2000.Phylogeny of the genus Pseudomonas: intragenic structure reconstructed fromthe nucleotide sequences of gyrB and rpoD genes. Microbiology 146, 2385-2394.
[13] Hilario, E., Buckley, T.R., Young, J.M., 2004. Improved resolution on the phylogenetic relationships among Pseudomonas by the combined analysis of atpD, carA, recA and 16S rDNA. Antoine van Leeuwenhoek 86, 51-64.
[14] Mulet, M., Lalucat, J., Garcìa-Valdes, E., 2010. DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513-1530.
小组成员及其分工
张俊:数据收集整理、软件安装与测试、程序代码编写、进化树构建及美化、博客园文本撰写整理
陈绅:博客园文本整理
钟晶莹:博客园文本整理
赵海芹:博客园文本整理