1 引言
测序技术能够为研究学者带来大量的测序数据,而测序技术的不断优化使得测序数据更加准确。从头测序是一项不依赖于任何已知或参考序列的测序技术,它利用生物信息学分析技术将序列片段进行拼接、组装以实现整个序列的鉴定,可用于未知基因组、转录组和蛋白质的全序列分析。
在蛋白质组学中,利用串联质谱对肽段序列进行分析和鉴定,然后推测样品中的蛋白质,这就是常用的高通量测序方法。其中,肽段是通过对蛋白质进行酶切产生的,数据分析则是利用串联质谱分析产生的二级图谱中的肽段序列信息来确定样品中的蛋白质。这在计算策略上可以分为从头测序方法、数据库搜索方法和肽段序列标签方法。为了弥补传统蛋白质鉴定方法在未知蛋白质序列和突变分析上的不足,促生了蛋白质从头测序这一新技术。蛋白质从头测序技术是指不依赖于现有的生物信息数据库,在图谱中依据肽段碎裂的规律推导出肽段的序列。
蛋白质从头测序的原理是在没有序列库进行比对的情况下,基于蛋白酶切后的肽段分子在质谱检测中有规律性的断裂,找到特定断裂模式,再根据质谱峰之间质量差即可算出对应的氨基酸信息,以及氨基酸上的翻译后修饰。从头测序最重要、最关键的就是对已测得的小片段进行拼接、组装,如果在这个过程中发生拼接错误,那么将会导致整个测序结果不准确。因此,在测序前将待测样品进行多重酶切以及对序列进行反向验证是保证片段全覆盖以及测序结果准确性的关键因素。
蛋白质组学研究的重点是大规模研究蛋白质组的特征,即生物体内的全部蛋白质。在蛋白质组学中,从串联质谱(tandemMS)数据中进行肽段从头测序在新蛋白质序列的表征中起着关键作用。在过去的20年里,这一领域得到了积极的研究,最近对单克隆抗体的“淘金热”无疑将从头测序的应用提升到了一个新的水平。
2 蛋白质从头测序技术的算法及方法
2.1 算法
基于质谱对蛋白质鉴定的算法可分为两大类:数据库检索和从头测序。数据库检索是将质谱数据与数据库进行对比和匹配获得肽段信息,从头测序不依赖蛋白质的数据库,直接通过谱图中的离子信息来推导肽段的序列。蛋白质从头测序技术可以对未知序列蛋白质、发生突变的蛋白质以及发生翻译后修饰的蛋白质进行测序。目前蛋白质从头测序的主要算法有PEAKS系列、p Novo系列、Pep Novo、Novor等。Muth等研究者评估了Novor、PEAKS和Pep Novo三种测序软件在实验和模拟数据集上测序的准确度和运行时间,这三软件虽然对模拟数据的测序准确度可达到84%,但对高分辨率的实验数据测序准确度最高只有35%。虽然目前发展的蛋白质从头测序算法较多且种类不断更新,但由于质谱谱图中存在着噪声和干扰离子,无法准确地识别谱图中肽段的特征碎片离子。精准解析谱图的难度增加且测序准确率较低,在肽段的从头测序准确率低的情况下,使用序列拼接软件对序列进行正确的拼接是有一定难度的。
2.2 方法
2.2.1 穷举法
利用穷举法对蛋白质肽段序列进行鉴定是对蛋白质从头测序方法的最初尝试。穷举法是列举出母离子质量误差范围内的所有可能的肽段,然后将候选肽段和实验串联质谱作比较,找出匹配度最高的候选肽段。对于长度相对较长的肽段序列,使用穷举法对长度较短的肽段序列进行从头测序带来的效果会更好,其计算速度也相对更快。
针对穷举法存在的问题,科研工作者提出了诸多方法来解决,最典型的解决方法是“剪枝方法”。剪枝方法是将测试质谱图中的碎片离子信息与理论质谱图中的碎片离子信息进行比较,挑选出符合条件的肽段序列,从而限制候选肽段的数量。即使该方法对肽段序列进行了筛选从而提高了候选肽段的质量,但仍然会出现误删正确的候选肽段的情况。
2.2.2 谱图模型
谱图模型是将串联质谱图中的信息进行整理并提取出来以构建有向无环图,通过求解有向无环图的最长路径来解决肽段的从头测序问题。
首先将所有观测到的碎片离子以质荷比为准则,按照从小到大的顺序进行排列,如果两个碎片离子相差一个氨基酸的质量,那么将这两个碎片离子作为图的节点,两个节之间的连线作为图的边并用相应的氨基酸进行表示,按照上述方法构建出一个有向无环图。由此,如何解决肽段的测序问题转化为找出经过该有向无环图中所有节点的最长路径。
在谱图模型中,串联质谱中的第i个碎片离子用si表示,则肽段对应的串联质谱用S={s1,s2,..}表示,用Mi和Ii.表示si的质荷比和离子强度,那么肽段母离子的质量则用M表示。当串联质谱中某个碎片离子是b系列碎片离子或者是y系列碎片离子时,其对应的肽段序列的前缀质量可以用bi或ti表示。bi和ti是有向无环图的两个顶点,而碎片离子si;则对应这两个顶点。不仅如此,这个有向无环图中所有的顶点都需要满足的条件:bi=Mi;或bi=M-Mi;且bi<ti,bi=M-ti;b0<b1<..<bn<tn<...<t1<t0。
在早期的谱图模型中,构建连接图前所采用的流程基本是一致的。随着技术的发展,研究者不断优化谱图模型。比如,Lutefisk方法是结合碎片离子的类型,利用质谱中强度较大的离子构建有向无环图,寻找该有向无环图的最长路径,从而获取蛋白质测序的结果。Sherenga方法则是以早期的谱图模型为基础给图中的每个节点增加一个权重,每个权重就是节点对应的碎片离子的质荷比。当两个节点之间的权重差值等于一个氨基酸的质量,那么就用该氨基酸对其进行标记。由此可见,谱图模型的优势在于其计算简单、便于理解,但也存在一些不容忽视的问题。当碎片离子的类型变多时,谱图模型的计算复杂度就会增加。而当碎片离子受到其他因素影响时,这会影响最长路径的求解结果,肽段测序结果的准确性就会降低。
3 蛋白质从头测序技术的应用
3.1 对牛奶、花生、鸡蛋、大豆中主要致敏蛋白的鉴定
食物过敏是易致敏人群摄入的某些食物中含有过敏原从而引发的一种不良反应,也被称为食物变态反应。在日常生活中,牛奶、花生、鸡蛋和大豆制品中的许多蛋白都是过敏原,影响和制约着致敏人群(特别是婴幼儿)的正常饮食与营养摄入。因此,在食品行业的加工生产过程中,食物致敏是食品研发和检测人员必须面对的问题。
刘焕龙利用Bottom-up MS 蛋白组学的思路,将牛奶、花生、鸡蛋、大豆样本中蛋白质进行提取,通过还原烷基化,利用碱性Trypsin酶切形成肽段,使用超高效液相色谱对混合多肽段进行色谱分离,联用静电场轨道阱高分辨质谱对混合多肽段进行数据采集和质谱分析,最终把采集到的质谱数据导入软件PEAKS Studio 8.5中,运用从头测序技术进行蛋白分析,从而对所有蛋白种类、致敏蛋白质种类进行筛选鉴定。
结果分别从牛奶中、花生、鸡蛋和大豆中识别和鉴定出了30、35、25和52种蛋白质以及其主要致敏蛋白质。根据四种食物中主要蛋白质的相对丰度统计结果表明牛奶、花生、鸡蛋和大豆的主要致敏原分别为Bos d 9、Ara h 1、Gal d 2和Gly m 6。
从头测序技术的运用,为建立相关致敏蛋白数据库等提供很好的技术支持,保障过敏人群的生命安全。
3.2 对多发性骨髓瘤患者的抗体轻链蛋白型测定
多发性骨髓瘤(MM)是一种浆细胞恶性肿瘤,在多发性骨髓瘤疾病中,单克隆免疫球蛋白轻链(LCs)大量产生,形成影响各种器官(如肾脏)的沉积物,同时保持可溶性,在等离子体中浓度高达几g·L–1。为了更好地了解影响LCs溶解度及其聚集倾向的因素,一个包含LCs生物物理特性及其序列的数据库至关重要。
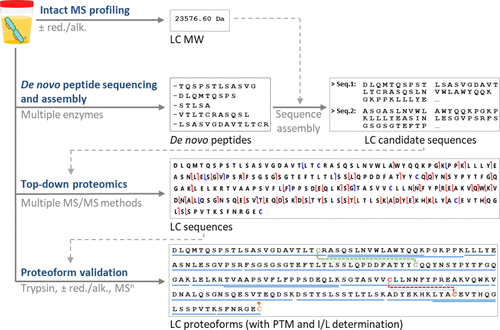
图1 从头测序策略
Mathieu Dupré等人通过结合完整质谱分析、使用多种酶的从头测序、TDP与多种MS/MS方法以及带或不带还原/烷基化的BUP,以可靠地鉴定LCs蛋白型(如图1)。基于上述技术,研究首次表征从患者尿液中提取的LCs在序列,PTM以及单体或二聚体形式的存在方面的高变异性。
从头测序技术的运用,帮助研究人员获得关于完整蛋白质型获得的数据,实现了确定影响LCs聚集倾向并导致疾病主要因素的目标,为后续了解聚合过程并能够在将来防止它提供思路。
4 蛋白质从头测序技术面临的挑战
蛋白质从头测序技术主要是对质谱图进行处理,每个图谱的从头序列都有可能包含测序错误,这也是基于串联质谱技术的蛋白质从头测序法的基本局限性。如果处理不当,肽段上的错误会延伸到最终的蛋白质测序结果中。降低从头测序在肽段上的错误率仍是目前需要解决的问题之一。同时,在从头测序的过程中,蛋白质经过特异性蛋白酶酶解后得到的肽段,因为有些肽段长度过短或肽段间序列重叠部分少,无法将这些肽段序列拼接成完整的蛋白质的序列。
已有研究发现,采用一种使用非特异性蛋白酶连续酶解的蛋白质测序方法可以解决上述问题。首先验证非特异性蛋白酶在不同酶解时间产生不同种类的肽段,使用从头测序对得到的肽段进行分解、叠加和延伸,最终可拼接成完整的蛋白质序列,完成蛋白质的序列测定。针对非特异性蛋白酶,分别使用菠萝蛋白酶、糜蛋白酶、弹性蛋白酶、蛋白酶K、链霉蛋白酶和嗜热菌蛋白酶对牛血清白蛋白(BSA)进行一次酶解。使用PEAKS(v8.5)软件对BSA经连续酶解产生的肽段从头测序,得到候选肽段序列。再将每种蛋白酶酶解后得到的候选序列与BSA序列进行匹配(如图2)。最后进行蛋白质序列拼接,使用序列拼接算法,将从头测序得到的肽段序列进行拼接(如图3)。
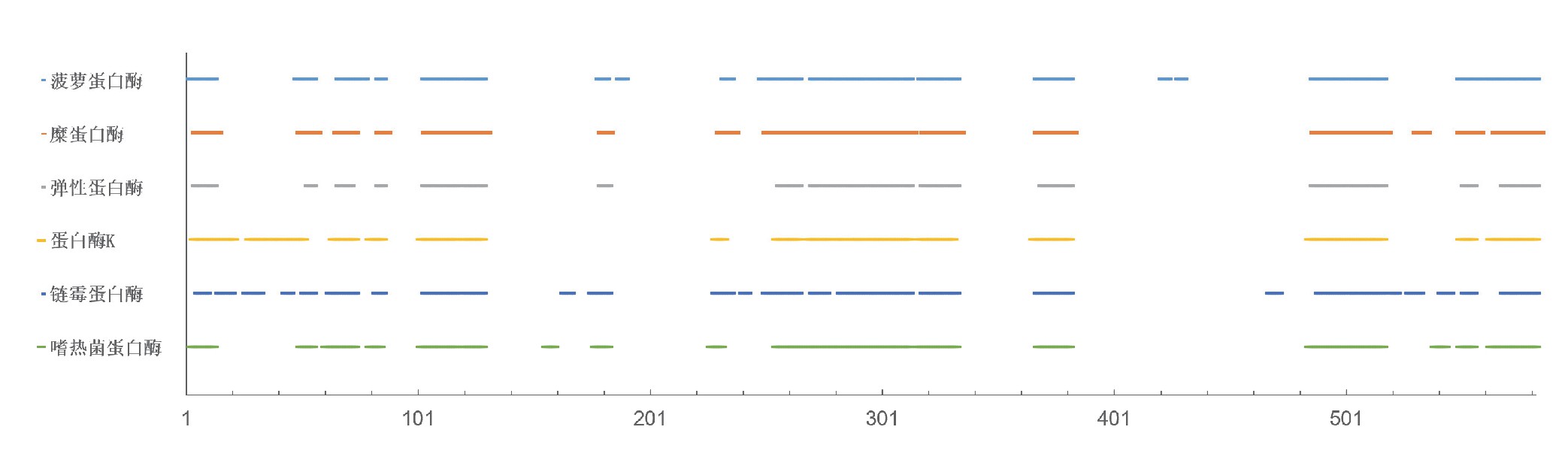
图2 从头测序肽段的序列覆盖度
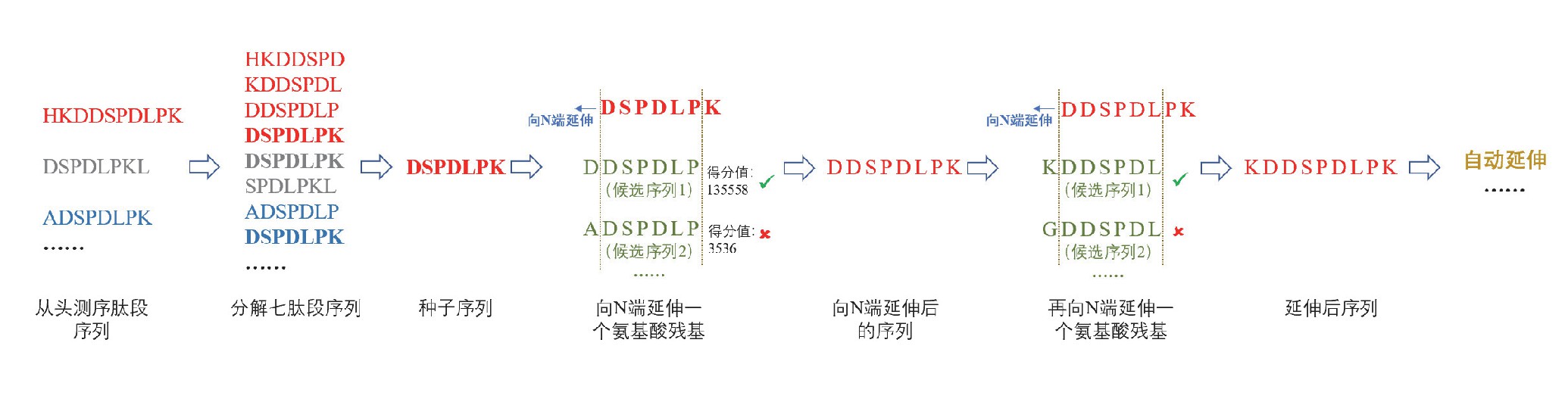
图3 序列拼接流程
同时,计算方面的挑战也仍然存在。因为MS/MS谱包含大量的噪声和模糊性,需要通过各种形式的动态规划进行严格的全局优化,已有研究将神经网络和深度学习引入到肽从头测序。在基因组学领域,深度神经网络模型已经被开发用于预测非编码单核苷酸变异的影响,预测蛋白质DNA和RNA结合位点,蛋白质接触图预测和MS成像。深度学习的关键方面是它通过多层神经元学习高维数据的多层表示的能力,与传统的机器学习方法不同,这些特征层不是基于特定领域的知识预先设计的,因此在发现数据的复杂结构方面具有更大的灵活性。
蛋白质从头测序技术在挑战中发展!
小组成员及分工
张玉君:负责引言、蛋白质从头测序技术算法及挑战部分
张畑钿:负责蛋白质从头测序技术方法部分
张嘉敏:负责蛋白质从头测序技术应用部分,博文整理汇总