对“美国出现的SARS-CoV-2的分子进化特征”一文的简要介绍
冠状病毒(CoV)是一种RNA病毒,可感染人类和其他哺乳动物,可引起多种疾病,例如呼吸道、肠道、肝脏和神经系统疾病。感染人类的冠状病毒有七种类型,包括SARS-CoV、MERS-CoV、HCoV-229E、HCoV-HKU1、HCoV-NL63、HCoV-OC43和SARS-CoV-2, SARS-CoV-2的基因组序列和刺突蛋白结构SARS-CoV相似。SARS-CoV-2是2019年底新发现的beta冠状病毒,具有高致病性,对人类健康构成严重威胁。截至2021年7月,全球SARS-CoV-2感染人数已达到1.8亿人。至于美国,已确诊超过3300万例病例,接近全球确诊病例总数的四分之一。美国的死亡人数超过6000万。有学者分析了SARS-CoV-2在疫情爆发的第一个月的全球进化率,估计每年每个位点平均核苷酸突变率范围为1.7926*10^-3至1.8266*10^-3的取代。疫情爆发四个月后,突变率变为3.95*10^-4每年每个核苷酸,几乎比SARS-CoV的突变率低7倍,比MERS-CoV的突变率低2倍。但是,到目前为止,还没有关于美国病例的相关研究报道,因此本文作者开展了本次研究。
受体结合和膜融合是SARS-CoV-2感染周期的初始和关键步骤,在此期间SARS-CoV-2刺突蛋白(S蛋白)起着关键作用。因此,研究SARS-CoV-2的S蛋白非常重要。本文作者以SARS-CoV-2全基因组序列和编码刺突蛋白(S基因)为研究对象进行生物信息学分析,研究其基因组和刺突蛋白的分子进化特征,采用MCMC方法计算全基因组序列的进化速率和S基因的核苷酸突变率。MCMC方法及马尔可夫链蒙特卡罗方法,是用来在概率空间,通过随机采样估算兴趣参数的后验分布。蒙特卡罗本身可以采样,马尔科夫链也可以进行采样,但是单独使用这两个方法需满足一是概率密度函数可积,第二个是累积分布函数有反函数,同时对于高维随机变量,容易出现“维数灾难”问题,所以将这两个方法进行结合使用。
首先是序列数据收集,作者采用了来自NCBI数据库的15个全基因组序列和相应的S基因序列,包括SARS-CoV-2, SARS-CoV, MERS-CoV, HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, 蝙蝠SARS-like, 果子狸SARS-CoV, 穿山甲CoV, 鼠肝炎病毒、大鼠CoV,刺猬CoV, 骆驼MERS-CoV, and 牛CoV,以及从NCBI病毒的严重急性呼吸综合征冠状病毒2数据中心获得的共计2241个全基因组序列和相应的S基因序列,再移除较低质量的序列后,剩余的1875个全基因组序列将被用于分析,并使用从Wuhan-Hu-1分离得到的MN908947作为参考序列。
在获得所需序列数据后,作者开始进行序列比对和系统动力学分析。利用软件MAFFT v7.464(MAFFT是常见的多序列比对工具之一,通常来说,它的比对速度弱于Muscle,但是准确性更强)对1875个SARS-CoV-2基因组序列进行多序列分析,利用软件ModelFinder(替代模型的选择是进行最大似然(ML)和贝叶斯(BI)分析前不可获缺的一个重要阶段。对于核酸苷替代模型的选择,常见的MrMTGUI(MrModelTest/ModelTest)配置复杂,不易操作;jModelTest 支持界面操作,但是耗用大量系统资源。因而IQ-Tree软件中的ModelFinder自然成了首选,最大的特点就是快)基于贝叶斯信息准则的结果为SARS-CoV-2基因组序列分析最优替代模型,最佳核苷酸取代模型的结果是GTR+F+G4。
突变率可以定义为每次细胞分裂、每代或每单位时间的突变数。作者采用马尔可夫链蒙特卡罗(MCMC)方法重构最大分支可信度树,并利用BEAST V2.6.2(Beast软件是采用贝叶斯演化分析的原理,用来估算系统发育关系和分化时间的软件,使用前提是需要有化石证据或其他的先验信息进行时间校准)计算突变率。为了设置数据集之前的时间尺度,使用了约束的进化速率,即对数正态先验平均值为10^-3每年每个站点的替代,使用松弛时钟对数-正态分子钟模型进行了系统发育贝叶斯分析,并根据相关研究选择了合并贝叶斯天际线作为种群规模和增长的模型。采用MCMC分别分析全基因组序列和编码刺突蛋白序列(S基因)的序列,计算突变率,长度为4*10^8steps,每4*10^4steps采样一次。所有参数(ESS>200,老化10%)的收敛性使用Tracerv1.7.1进行了验证。最终的MCC树由TreeAnnotator(BEAST2中的软件包)生成,并显示在Figtree v1.4.4中。
通过利用PROVEAN可以预测S蛋白的非同义突变是否会影响其功能,可以检测氨基酸的有害取代,并且预测取代是否会影响其表型。如果PROVEAN的分数不大于−2.5,则表示氨基酸替代有害,反之,如果分数高于−2.5则认为是中性。
作者在三个方面对结果进行讨论,第一个方面是SARS-CoV-2的突变率。根据BEAST2的结果显示,作者获得了每年每个站点6.677×10^−4的突变率(密度在95%是最高[HPD]: 6.117×10^−4——7.270×10^−4),对于全基因组,每年每个站点为8.066×10−4(95% HPD: 5.969×10−4——1.038×10−3)用于编码S蛋白质的序列。科学家发现的第一个更具传染性的突变是D614G,这是由于在23403位置的碱基A变为G而引起的。在69 098个氨基酸序列中发现了12个S基因的高频突变,在氨基酸(AA614)位置的突变频率最高,为98.47%(图1)。
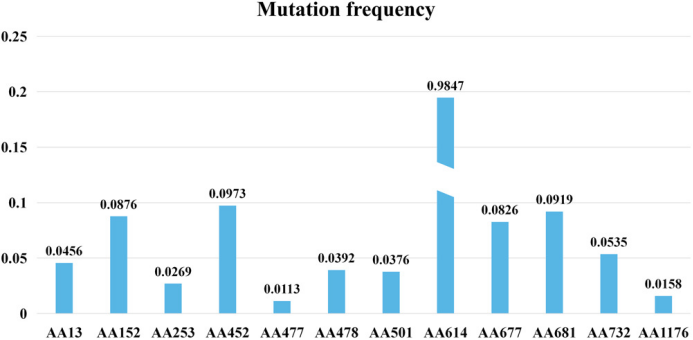
图1S蛋白突变的分布。S蛋白中的所有突变都是非同义的,位置AA614的频率最高
而图2显示了SARS-CoV-2分离株的贝叶斯系统地理重建的MCC树。为了使图像视觉效果更清晰而又不失基因组的代表性,我们在图2中仅显示380个基因组序列来显示MCC树。
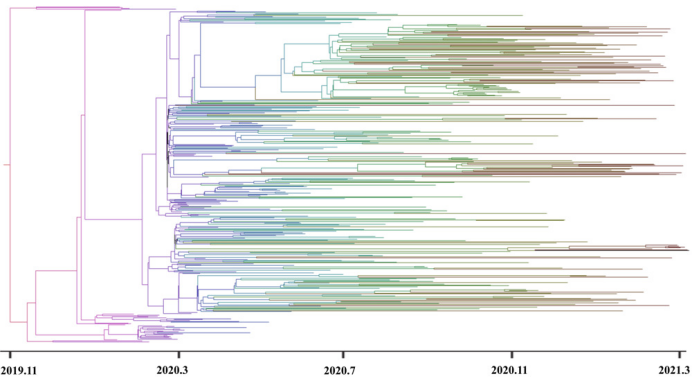
图2贝叶斯最大分支可信度树
作者估计,2019年11月5日是SARS-CoV- 2在美国出现的一个共同祖先最近的时间,HDP从2019年9月21日到2019年12月16日都持续在95%。这一结论也与2019年12月1日文献报道的第一例病例相一致。
第二个方面是SARS-CoV-2的系统发育分析,为探索SARS-CoV-2的进化选择压力,作者以MN908947为参考序列进行系统发育分析,将MN908947的全基因组与其他14种冠状病毒进行比对,建立系统发育树。以MN908947作为前景分支,其他14种冠状病毒作为分支位点模型的背景分支。结果显示,在全基因组和S基因中分别检测到13个和2个具有统计学意义的阳性位点。作者采用了RDP4和SimPlot检测15种冠状病毒的重组事件。然而,结果显示,并没有发现具有统计学意义的重组事件。
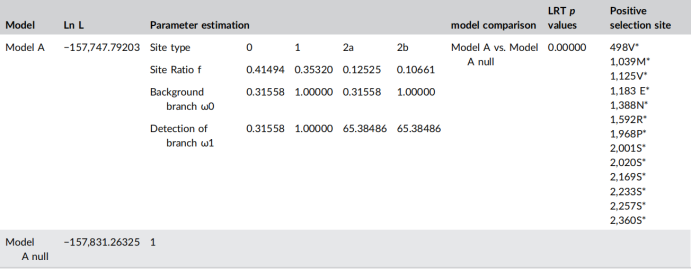
表1 SARS-CoV-2分支站点模型的结果
第三个方面,是S蛋白氨基酸的有害突变检测。作者发现在所有69 098个蛋白序列中有101个非同义突变。结果显示,有7个有害突变,其余94个均为中性突变。
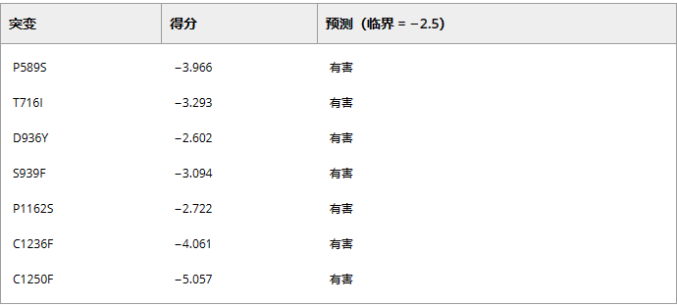
表2在S蛋白中检测到七个有害突变
SARS-CoV-2已经在世界范围内进化成了数千种变种。虽然这种病毒可能发生大量突变,但只有少数可能对人类造成严重伤害。与其他冠状病毒和RNA病毒相比,SARS-CoV-2的突变率(6.677×10^−4) 处于中等级别。此外,在感染过程中,突变率会逐渐降低。造成这种情况的原因可能是SARS-CoV-2有自己的拷贝“校对机制”,可以纠正复制过程中可能出现的一些错误,导致SARS-CoV-2突变率下降。另一方面,有害突变可能导致一定程度的蛋白质结构和功能变化,最终影响SARS-CoV-2的繁殖。在自然选择的压力下,随着有害突变的积累,病毒数量会逐渐减少,甚至可能发生突变崩溃,导致种群灭绝。
此外,在S蛋白的许多样本中发现了非同义突变,有害突变比例为6.93%。这些有害突变可能会影响S蛋白的结构和功能。一旦S蛋白RBD的氨基酸发生突变,与人结合的ACE2的受体亲和力可能会发生变化,这可能导致人类感染能力的增强,也使现有疫苗无效。从GISAID数据库中收集的全球突变数据显示,S基因的几乎每个位点都发生了突变。
病毒在每个复制周期中以不可预测的方式变异和重组来创建新的变种。从生物进化的角度来看,病毒的存在是人类的自然选择压力。人体的免疫反应会产生一定的适应性,这将促进人类向更有利于人类生存的方向发展。然而,人体免疫系统的进化和各种干预措施也可能促进病毒的适应性突变。通常,病毒在同一宿主中传播时会变得“温和”,因为高致病性会导致宿主死亡,导致传播和繁殖的丧失,不利于病毒本身的生存和繁殖。
实验结果表明,SARS-CoV-2毒株的一些变异株提高了感染性,如D614G、S477N和N439K等变异株,这些变异株比原病毒具有更高的传播能力和更快的复制速度,但致病性并未增加。
目前,世界上出现的SARS-CoV-2传染性较多的变种,包括第一个D614G变种B.1.5-B.1.72、英国变种B.1.1.7、南非变种B.1.351、巴西变种P.1和菲律宾变种P.3,以及美国的两个本土变种,加利福尼亚变种B.1.429/B.1.427和纽约变种B.1.526。新变种的传播能力、致病性和免疫逃避能力一直在不断地增加。其中,英国变种B.1.1.7是一种对人体影响较大的病毒。在2020年12月,新菌株B.1.1.7在英国被首次发现,其S基因序列累积了16个核苷酸突变,导致10个氨基酸位点改变(H69del、V70del、Y144del、N501Y、A570D、D614G、P681H、T716I、S982A、D1118H)。根据PANGO谱系的数据,B.1.1.7突变株已在90多个国家被发现。研究表明,该突变菌株的传染性比当时流行的菌株高50%以上。截至10月20日,该菌株已在美国造成20915例确诊感染,并已成为美国的主要传播类型。在2020年7月发现的加州突变体 B.1.427/B.2020.452 具有一个 L20R 突变位点,导致其传染能力增加了20%,并表现出中度的免疫逃逸,导致相当快的传播速率。2020年11月发现的纽约变种 B.484.477,其突变位点为 E484K或S477N,也表现出了中度的免疫逃逸。
总而言之,作者阐明了SARS-CoV-2在美国的进化特征,为未来监测和预防病毒变异提供了科学依据。为了控制SARS-CoV-2,有必要继续监测特定的突变,这对于进一步深入研究SARS-CoV-2和评估疫苗的有效性仍然具有重要意义。
参考文献:
1.Shihang Wang, Xuanyu Xu, Cai Wei, Sicong Li, Jingying Zhao, Yin Zheng, Xiaoyu Liu, Xiaomin Zeng, Wenliang Yuan, Sihua Peng,Molecular evolutionary characteristics of SARS‐CoV‐2 emerging in the United States
分工:吴梓伟、郭林丛:文献翻译
厉燕瑾:PPT制作
吴梓柯:汇报