2023年9月29日,哈尔滨工业大学王亚东团队联合中国农科院韩天富团队在bioRxiv发表了题为“A telomere-to-telomere genome assembly of Zhonghuang 13, a widely-grown soybean variety from the original center of Glycine max”的研究文章,该文于11月3日正式见刊《The Crop Journal》。

主要结果
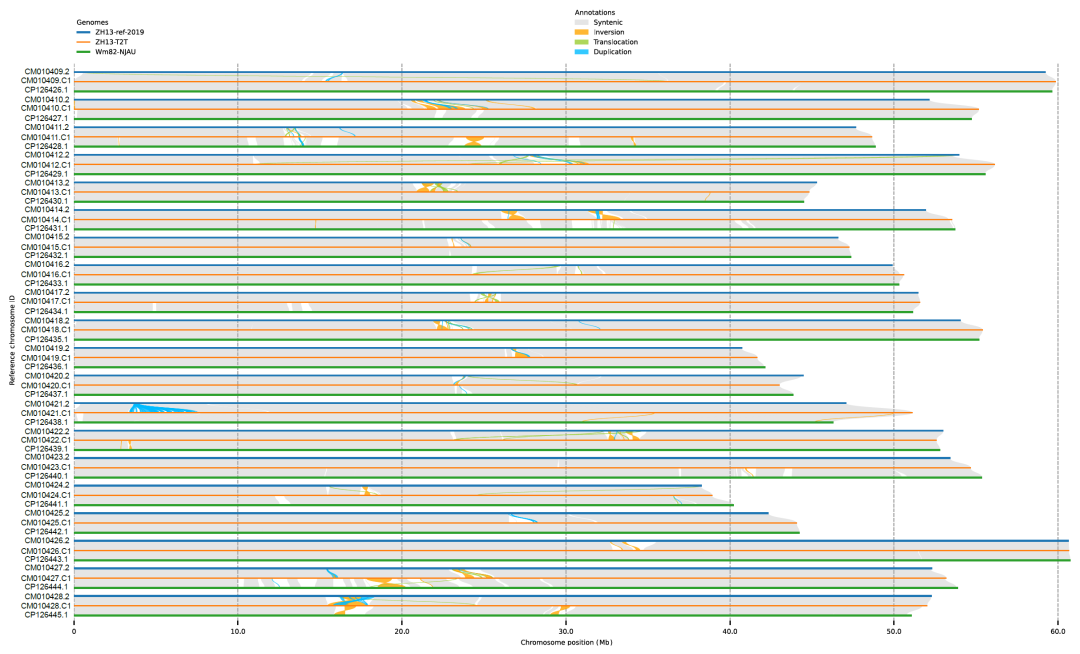
相比之前发表的中黄13基因组,ZH13-T2T填补了全部393个空白区域。拼接产生的基因组长度为1,015,024,879 bp,N50长度为52,033,905 bp。
20条染色体中的全部40个端粒区域均成功拼接完成,端粒中位数长度达到8449 bp;BUSCO指标达到99.8%,碱基质量评分(Merqury质量评分)达到46.441。
PacBio Hifi和ONT超长测序数据在全基因组上的覆盖度均匀且符合期望值,未发现覆盖度异常区域,显示整个基因组具有较高的拼接质量。
ZH13-T2T基因组与原ZH13比较:共有421个长度超过5kb的显著序列差异区域(总长16.3 Mbp)及112个结构变异,包括30个倒置变异、15个易位变异、67个序列重复变异。
ZH13-T2T基因组与南京农业大学最新发表的Williams 82 T2T基因组(Wm82-NJAU)进行比较:发现了162个长度超过5 kb的显著序列差异区域(总长23.02 Mb)和30个结构变异(包括16个倒位变异、7个易位变异和7个序列重复变异)。
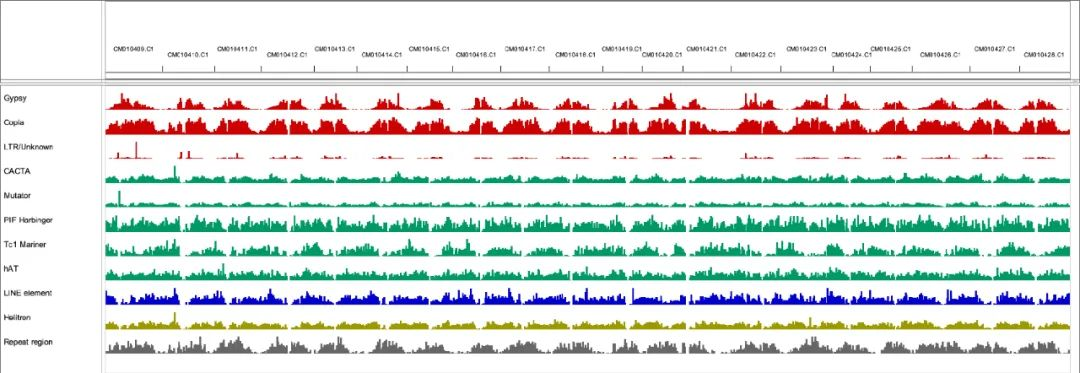
ZH13-T2T重复序列注释,发现了大量具有高置信度的新编码基因和重复元件(57.07%),其中反转录转座子占38.16%(包括0.12%的SINEs、1.58%的LINEs和36.47%的LTR元件),而DNA转座子占6.72%。串联重复序列占大豆基因组的2.63%(26.65 Mb),明显超过了在中黄13参考基因组序列中观察到的1.03%(10.54Mb)。
ZH13-T2T基因结构注释,共获得50,564个高置信度蛋白质编码基因。与ZH13-2019相比,在gap区域内发现了707个新基因。在此前的gap区域中,在CM010421. C1染色体的14.84-17.73Mb区域内观察到新发现的基因数量最多,共135个新基因。此外,在这些gap区域内确定了42,668个TEs,300个GmCent-1元素和586个GmCent-2元素。RNA-seq分析显示,在38个gap区域中的295个基因中有表达。
染色体着丝粒鉴定:通过串联重复查找工具(TRF)鉴定ZH13-T2T基因组中可能构成着丝粒的重复单体,发现长度为91 bp和92 bp的大量串联重复序列,与着丝粒区域TE的间隙相吻合。20个着丝粒的平均长度为2.40 Mb,在CM010410.C1染色体上观察到的最长着丝粒(4.42 Mb) 和在CM010421.C1染色体上观察到最短的着丝粒(0.66 Mb)。
着丝粒长度与染色体大小之间没有显著相关性。着丝粒的相对位置在不同染色体之间存在差异,最小L/S比(长臂长/短臂长)为1.02(CM010415.C1),最大L/S比为2.95(CM010423.C1)。在ZH13-T2T的着丝粒区共鉴定出8个基因,主要富集染色质DNA结合、mRNA顺式剪接、组蛋白结合、基底转录因子、剪接体和嘧啶代谢。
着丝粒序列由96.0%的着丝粒卫星DNA(CentC)、着丝粒反转录转座子(CRM)和其他非CRM Gypsy反转录转座子组成。这些成分的比例在不同的着丝粒中差异很大,GmCent-1从0.0%到73.3%,GmCent-2从0.0%到90.4%,CRM从0.0%到2.2%,其他非CRM Gypsy反转座子从7.3%到68.2%不等。几乎所有的着丝粒都富含 CentC。
主要方法
数据策略
- PacBio Hifi测序(数据量:96.89 Gbp)
- ONT超长测序(数据量:96.63 Gbp)
- Illumina全基因组测序(数据量:55.40Gbp)
- Illumina Hi-C测序(数据量:106.4 Gbp)
工具
Hifiasm、NextDenovo和Canu的组装:
-
Hifiasm的contigs作为骨架(并比对到ZH13-2019版本进行排布定向),因为其准确性和连续性。
-
NextDenovo的contigs用于填充未解决的区域,而Canu的contigs用于填充gap、LCR校正和端粒细化。
-
另外,还做了很多组装微调的工作:通过监测读长覆盖度(高覆盖区HCRs和低覆盖区LCRs)来防止错误组装,并使用局部读长覆盖度进行质量控制。通过收集和对齐锚定的reads,来推断长重复序列。
端粒的鉴定:
-
使用7-mer重复序列(CCCTAAA / TTTAGGG)来识别初步组装中的端粒。
-
使用7-mer模式搜索由NextDenovo、Canu和Hifiasm(仅使用hifi reads)产生的contigs,并找到了其中的三个缺失。
-
使用李恒大神的seqtk确定了在ZH13-T2T基因组中端粒的精确位置,命令
seqtk telo -s 1 -m CCCTAAA ref.fa。
着丝粒的鉴定:
- 利用两个高拷贝的卫星重复亚家族CentGm-1和CentGm-2(它们专属于中心区域),确定大致位置。然后通过IGV观察到TE注释缺失的区域与91/92bp长序列集中的区域之间存在重叠,从而确定中心粒的位置。
结构变异鉴定:
- minimap2+SyRI
问题来了
今年8月底,南农宋庆鑫团队在MP上发表了Williams 82的T2T基因组。10月6号,广西大学王海峰和与东北农大陈庆山团队在MP发表了中黄13的T2T基因组和表观遗传修饰图谱。
本研究在9月29日投稿预印版,报道称“首个无间隙的中国大豆品种T2T基因组”。这就有点意思了,你认为哪个是首个大豆T2T?哪个是首个中国大豆T2T呢?
撞车的事情时有发生,竞争如此激烈。不过,纯基因组组装发中科院一区,还要什么自行车。
更多信息请关注微信公众号:生物信息与育种
