
#1,我们先下载毛果杨的基因组文件和GFF注释文件,自己去NCBI下:(https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000002775.5/),选Genbank的。
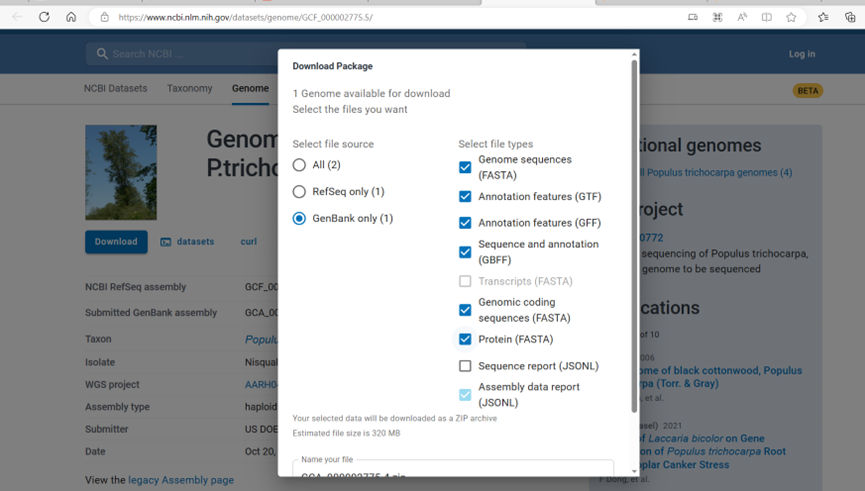
#2,我们将GFF文件和genomic.fna文件上传到服务器,并重命名下,Ptri_genome.gff和Ptri_genome.fa。
#3,安装hisat2
conda install hisat2
#4,对基因组构建索引,大概15min
hisat2-build Ptri_genome.fa genome
#5,生成批量的hisat2命令:
awk '{print "nohup hisat2 -x genome -1 "$1".clean.fq.gz -2 "$2".clean.fq.gz -S "$3".sam > "$3".temp.txt 2>&1 &"}' sample.txt >command_hisat2.sh
#6,查看command_hisat2.sh文件
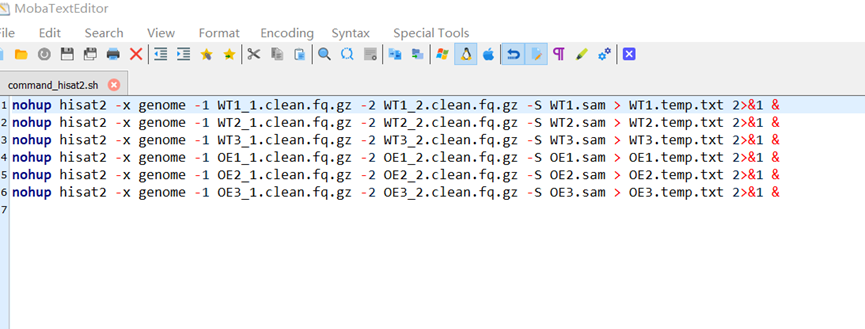
#7,运行,将转录组比对到基因组上,文件已经有挂载的,估计得好几个小时。
sh command_hisat2.sh
#8,安装samtools软件
conda install samtools
#检查软件是否可用,若有报错请看教程(https://www.cnblogs.com/nkwy2012/p/8986106.html)(https://zhuanlan.zhihu.com/p/408869413)
samtools
#9,生成批量的samtools命令
awk '{print "samtools view -bS "$3".sam > "$3".bam &"}' sample.txt >command_samtobam.sh
#10,查看command_samtobam.sh
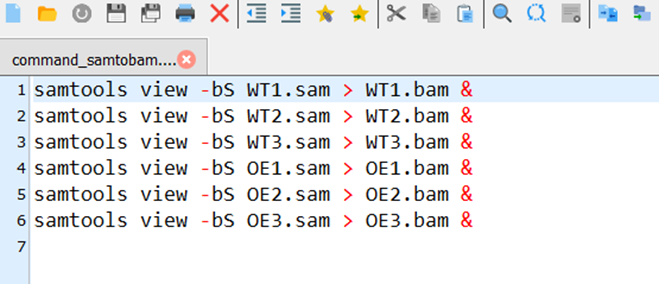
#11,运行,约1h。
nohup sh command_samtobam.sh &
#12,生成批量的sort.bam文件命令
awk '{print "samtools sort -o "$3".sort.bam "$3".bam &"}' sample.txt >command_sort.sh
#13,检查command_sort.sh

#14,运行,约30min。
sh command_sort.sh
#15,最终得到了sort.bam文件,共6个,sam和bam文件删掉,只留sort.bam文件。
#16,或者从步骤5开始使用for循环:
for i in {1..3};do hisat2 -x genome -1 WT${i}_1.clean.fq.gz -2 WT${i}_2.clean.fq.gz -S WT${i}.sam;done; for i in {1..3};do hisat2 -x genome -1 OE${i}_1.clean.fq.gz -2 OE${i}_2.clean.fq.gz -S OE${i}.sam;done; for i in {1..3};do samtools view -bS WT${i}.sam > WT${i}.bam;done; for i in {1..3};do samtools view -bS OE${i}.sam > OE${i}.bam;done; for i in {1..3};do samtools sort -o WT${i}.sort.bam WT${i}.bam;done; for i in {1..3};do samtools sort -o OE${i}.sort.bam OE${i}.bam;done;
#钢之炼金术师