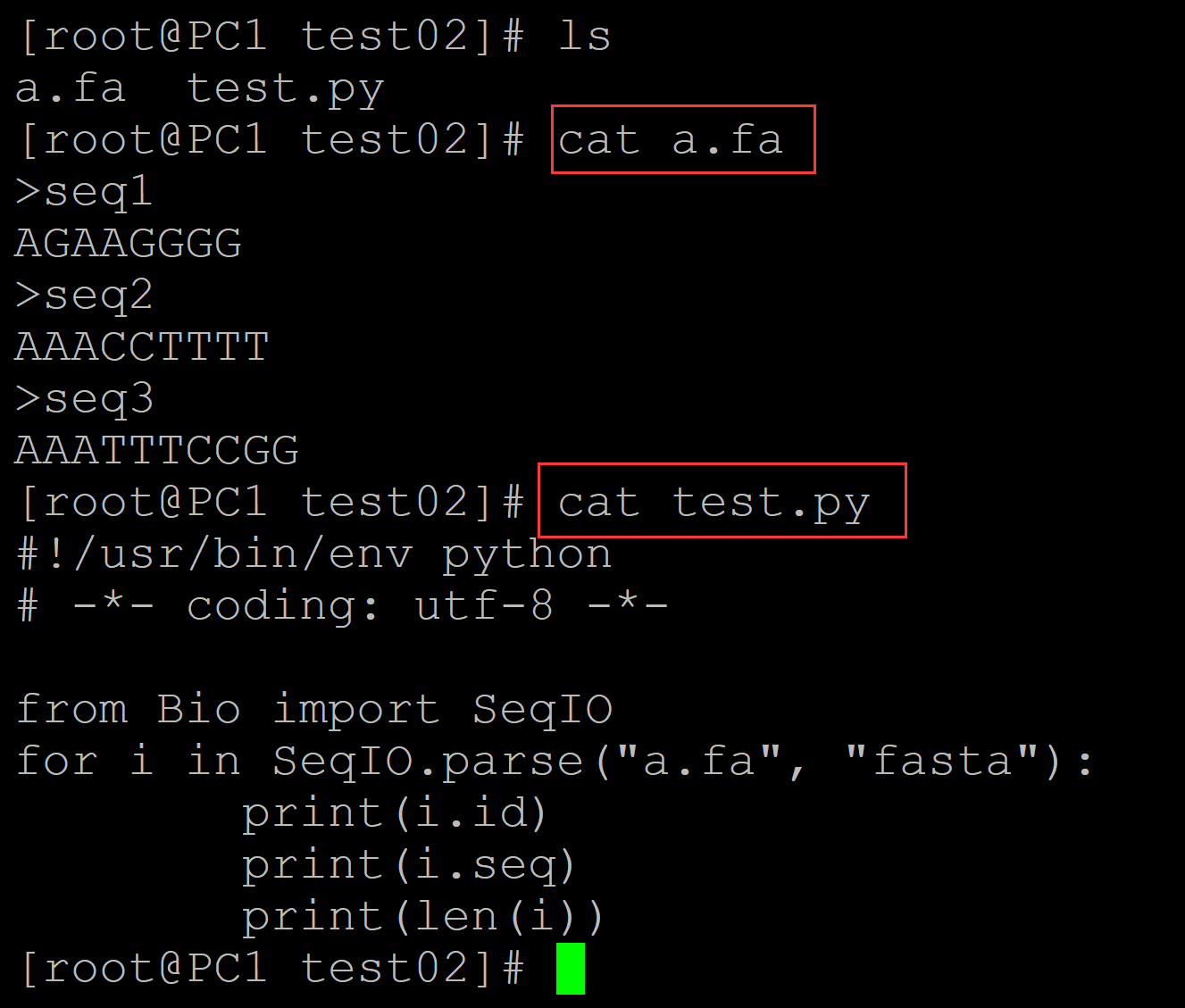
001、分别输出染色体ID、序列和序列的长度
[root@PC1 test02]# ls a.fa test.py [root@PC1 test02]# cat a.fa ## 测试数据 >seq1 AGAAGGGG >seq2 AAACCTTTT >seq3 AAATTTCCGG [root@PC1 test02]# cat test.py ## 程序 #!/usr/bin/env python # -*- coding: utf-8 -*- from Bio import SeqIO for i in SeqIO.parse("a.fa", "fasta"): print(i.id) print(i.seq) print(len(i))
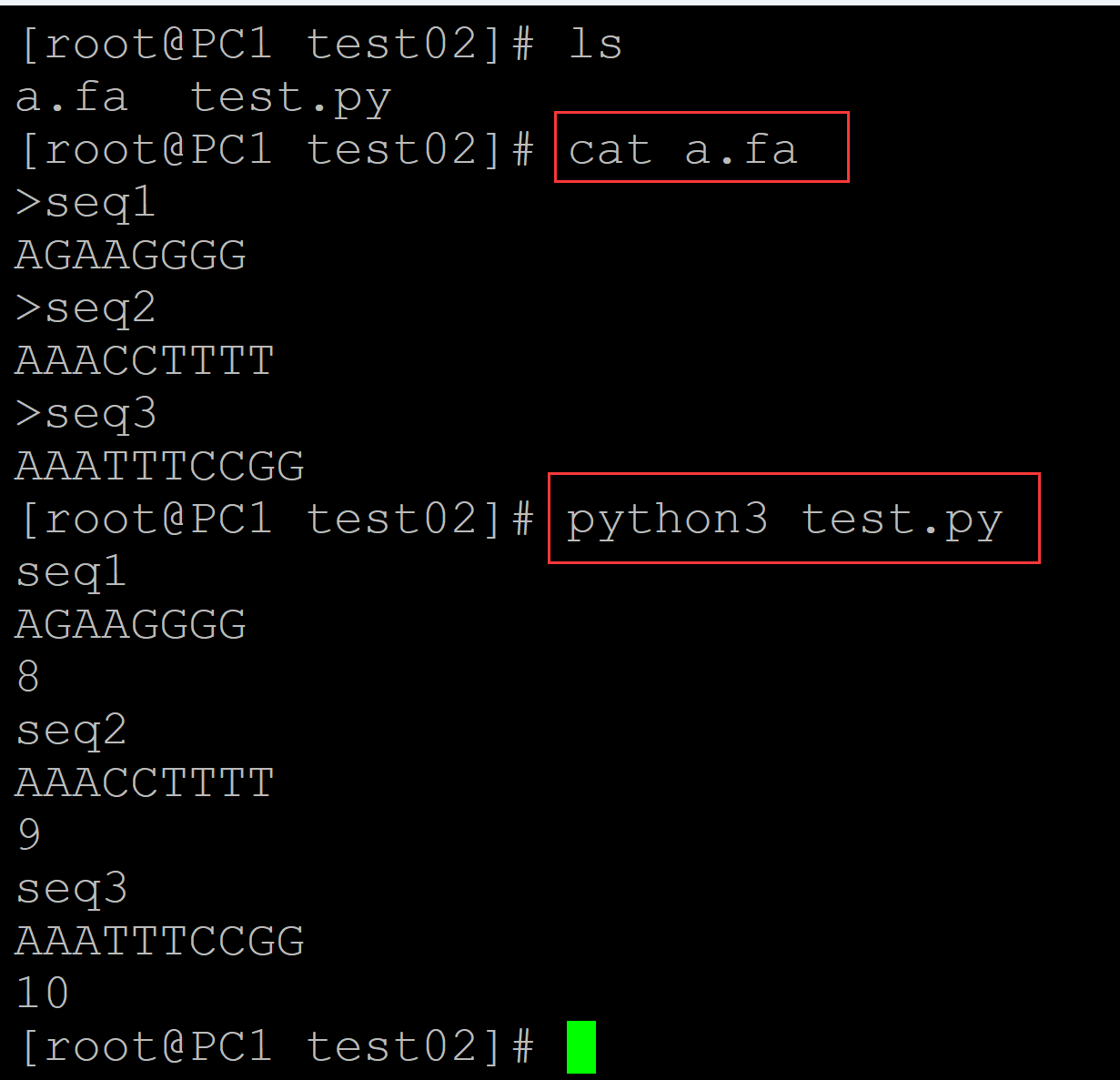
002、测试程序效果
[root@PC1 test02]# ls a.fa test.py [root@PC1 test02]# cat a.fa ## 测试程序 >seq1 AGAAGGGG >seq2 AAACCTTTT >seq3 AAATTTCCGG [root@PC1 test02]# python3 test.py ## 执行程序, 分别输出染色体ID、序列和序列的长度 seq1 AGAAGGGG 8 seq2 AAACCTTTT 9 seq3 AAATTTCCGG 10
。