写在前面
学习一个软件最好的方法就是啃它的官方文档。本着自己学习、分享他人的态度,分享官方文档的中文教程。软件可能随时更新,建议配合官方文档一起阅读。推荐先按顺序阅读往期内容:
文献篇:
1.文献阅读:(Seurat V1) 单细胞基因表达数据的空间重建
2.文献阅读:(Seurat V2) 整合跨越不同条件、技术、物种的单细胞转录组数据
3.文献阅读:(Seurat V3) 单细胞数据综合整合
4.文献阅读:(Seurat V4) 整合分析多模态单细胞数据
5.文献阅读:(Seurat V5) 用于集成、多模态和可扩展单细胞分析的字典学习
教程篇:
1.Seurat Tutorial 1:常见分析工作流程,基于 PBMC 3K 数据集
2.Seurat Tutorial 2:使用 Seurat 分析多模态数据
3.Seurat Tutorial 3:scRNA-seq 整合分析介绍
4.Seurat Tutorial 4:映射和注释查询数据集
5.Seurat Tutorial 5:使用 reciprocal PCA (RPCA) 快速整合
官网教程:
https://satijalab.org/seurat/articles/integration_large_datasets
对于非常大的数据集,标准的整合工作流程有时会在计算上非常昂贵。在此工作流程中,我们采用了两个可以提高效率和运行时间的选项:
- Reciprocal PCA (RPCA)
- Reference-based integration
主要的效率改进在 FindIntegrationAnchors()。首先,我们使用 reciprocal PCA (RPCA) 而不是 CCA,来确定一个有效的空间来寻找 anchors。当使用 reciprocal PCA 确定任意两个数据集之间的 anchors 时,我们将每个数据集投影到其他 PCA 空间中,并通过相同的相互邻域要求约束 anchors。所有下游整合步骤保持不变,我们能够“correct”(or harmonize)数据集。
此外,我们使用 reference-based integration。在标准工作流程中,我们识别所有数据集对之间的 anchors。虽然这在下游整合中赋予了数据集同等的权重,但它也可能成为计算密集型的。例如,当整合 10 个不同的数据集时,我们执行 45 种不同的成对比较。作为替代方案,我们在此介绍了将一个或多个数据集指定为综合分析的“reference”,其余指定为“query”数据集的可能性。在此工作流程中,我们不识别成对 query datasets 之间的 anchors,从而减少了比较次数。例如,当将 10 个数据集与一个指定为 reference 的数据集整合时,我们仅执行 9 次比较。Reference-based integration 可以应用于 log-normalized 或 SCTransform-normalized datasets。
此替代工作流程包括以下步骤:
- 创建要集成的 Seurat 对象列表
- 对每个数据集分别执行归一化、特征选择和标准化
- 对列表中的每个对象运行 PCA
- 整合数据集,进行联合分析
总的来说,我们观察到标准工作流程与此处演示的工作流程之间惊人相似的结果,计算时间和内存都大大减少。但是,如果数据集高度不同(例如,跨模态映射或跨物种映射),其中只有一小部分特征可用于促进整合,您可能会使用 CCA 观察到更好的结果。
对于这个例子,我们将使用来自人类细胞图谱的“Immune Cell Atlas”数据。
library(Seurat)
获取数据后,我们首先进行标准归一化和变量特征选择。
bm280k.data <- Read10X_h5("../data/ica_bone_marrow_h5.h5")
bm280k <- CreateSeuratObject(counts = bm280k.data, min.cells = 100, min.features = 500)
bm280k.list <- SplitObject(bm280k, split.by = "orig.ident")
bm280k.list <- lapply(X = bm280k.list, FUN = function(x) {
x <- NormalizeData(x, verbose = FALSE)
x <- FindVariableFeatures(x, verbose = FALSE)
})
接下来,选择下游整合的功能,并在列表中的每个对象上运行 PCA,这是运行替代 reciprocal PCA 工作流所必需的。
features <- SelectIntegrationFeatures(object.list = bm280k.list)
bm280k.list <- lapply(X = bm280k.list, FUN = function(x) {
x <- ScaleData(x, features = features, verbose = FALSE)
x <- RunPCA(x, features = features, verbose = FALSE)
})
由于此数据集包含男性和女性,我们将选择一名男性和一名女性(BM1 和 BM2)用于基于参考的工作流程。我们通过检查 XIST 基因的表达来确定供体性别。
anchors <- FindIntegrationAnchors(object.list = bm280k.list, reference = c(1, 2), reduction = "rpca", dims = 1:50)
bm280k.integrated <- IntegrateData(anchorset = anchors, dims = 1:50)
bm280k.integrated <- ScaleData(bm280k.integrated, verbose = FALSE)
bm280k.integrated <- RunPCA(bm280k.integrated, verbose = FALSE)
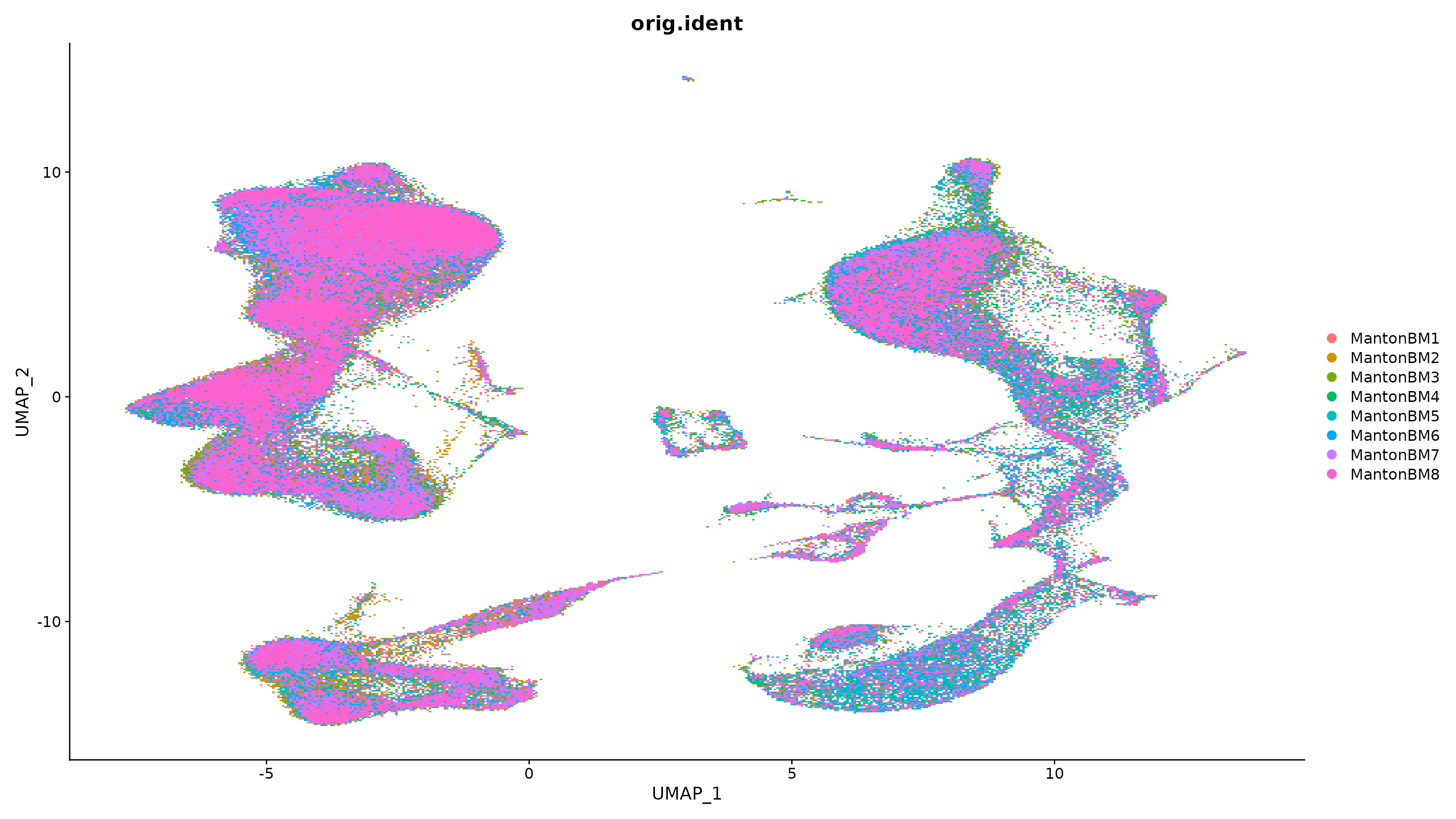
bm280k.integrated <- RunUMAP(bm280k.integrated, dims = 1:50)
DimPlot(bm280k.integrated, group.by = "orig.ident")

本文由mdnice多平台发布