利用代码复现一个机器学习应用于基因组预测的项目,张志武老师于2023年发表在《园艺研究》上的一篇文章。

目的
使用 GWAS 和 GP 结合重测序数据和从世界各地收集的 220 份紫花苜蓿种质的秋季休眠(Fall dormancy, FD),定义为秋剪后 30 天的植物再生高度:(i)比较不同GP方法的预测准确性;(ii)使用GWAS和GP的混合模型计算FD预测精度;(iii)扩展GP模型以进行新的性状预测。
示例代码
表型文件预处理
2017年温室种植,2018年移植大田,按三个重复的完全随机区组试验设计。2018年割茬三次,2019年割茬四次。秋季休眠是最后一次割茬后1个月的植物再生高度。使用广义线性模型 (GLM) 估计秋季休眠的方差:

广义遗传力计算:
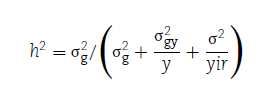
表型文件整理格式如下:

基因型文件预处理
#过滤保留双等位基因
vcftools --vcf mdp_genotype.vcf --min-alleles 2 --max-alleles 2 --maf 0.05 --recode --recode-INFO-all --out snp_maf0.05_allele2.vcf
#通过LD过滤
plink --vcf snp_maf0.05_allele2.vcf --indep-pairwise 100 50 0.2 --out snp_LD_filter.vcf --allow-extra-chr --make-bed
#挑选LD保存的标记,生成过滤后标记
plink --vcf snp_LD_filter.vcf --extract snp_filter.vcf.prune.in --out prunData --recode 12 --allow-extra-chr
#转换回vcf文件
perl /mnt/e/GWAS_model_test/tassel_v5/run_pipeline.pl -fork1 -plink -ped prunData.ped -map prunData.map -export snp_use_LD.vcf -exportType VCF -runfork1
#利用beagle软件填充缺失值
java -Xmx5g -jar beagle.22Jul22.46e.jar nthreads=20 gt=snp_use_LD.vcf out=snp_use_impute_out
#解压
gzip -d snp_use_impute_out.vcf.gz
#改成数值型格式,把0|0改成0,0|1改成1,1|1改成2,直接sed也行
perl change_vcf_format_impute.pl snp_use_impute_out.vcf > snp_numeric_format.vcf
# #去掉不需要的列
cut -f3,10- snp_numeric_format.vcf > R_use_format.vcf
#去掉注释信息,生成模型构建需要用的文件
grep -v '^#' R_use_format.vcf >R_use_format1.vcf
预处理后基因型文件格式如下:

机器学习建模预测
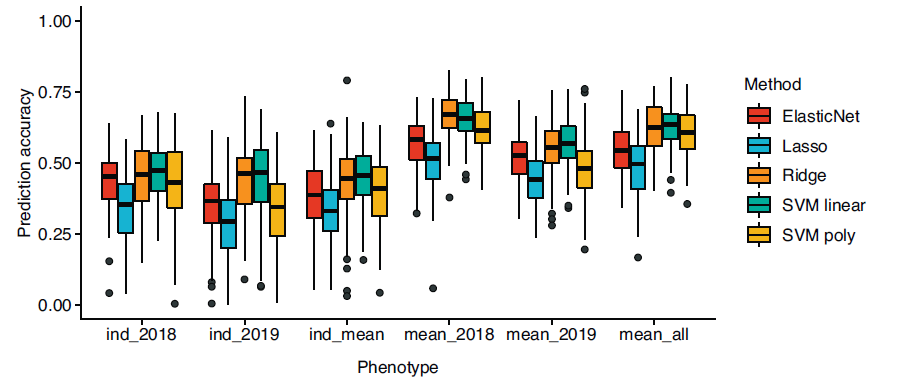
使用Python 包 sklearn 用于构建两类模型,一是SVM回归,包括线性核SVM linear和多核SVM poly;二是三种正则化模型,包括Lasso(L1正则化)、ridge regression(L2正则化)和 ElasticNet 模型 (L1和L2正则化结合)。
选择使用的5种模型分别代表了控制表型的不同QTN模型,Lasso代表少量大效应的QTN模型,Ridge代表大量微效QTN, ElasticNet代表Lasso和Ridge的中间类型。SVM(support-vector machines)是一种有监督的机器学习模型,从多纬度层面评估不同QTN间关系,分别从线性和非线性层面模型构建。
交叉验证方法:80%个体建模,20%个体作为预测,重复100次。代码如下:
#加载数据处理模块
import numpy as np
import pandas as pd
import scipy as sp
import statsmodels.api as sm
#加载机器学习模块
from sklearn import datasets, linear_model
from sklearn.linear_model import Ridge
from sklearn.kernel_ridge import KernelRidge
from sklearn.linear_model import ElasticNet
from sklearn.cross_decomposition import PLSRegression
from sklearn.metrics import mean_squared_error, r2_score
from sklearn.svm import SVR
from sklearn.ensemble import RandomForestRegressor
from sklearn.linear_model import ElasticNetCV
#导入基因型数据
data = pd.read_csv('R_use_format1.vcf',sep="\t",index_col=0)
#行列转置
data1=data.T
#导入表型数据
phe = pd.read_csv('pheno_all_ind_mean.txt',sep="\t",index_col=0)
#提取个体名称
index=phe.index
#匹配表型和基因型数据的个体信息
data1 = data1.reindex(index = index)
#挑选需要的表型
use_phe=phe["dormancy_ind_mean_all"]
#去掉表型为NA的个体
use_phe=use_phe[use_phe.notna()]
#把基因型数据也去掉对应的个体
GD1=data1.loc[use_phe.index]
#定义一个导出数据的文件
data_results={}
for i in range(100):
train_y = use_phe.sample(frac =.8) #拆分出80%个体为建模群体
train_x = GD1.loc[train_y.index] #得到对应的80%基因型数据
test_y = use_phe.drop(train_y.index,axis=0) #剩下20%个体作为验证群体
test_x = GD1.drop(train_y.index,axis=0) #20%个体对应的基因型数据
##以下模型可更换,去掉注释,选择其一
regr = Ridge(alpha=0.1) #岭回归模型
#regr = linear_model.Lasso(alpha=0.1) #lasso模型
#regr = ElasticNet(random_state=0) #ElasticNet模型
#regr = KernelRidge(alpha=1.0) #KernelRidge模型
#regr = PLSRegression(n_components=2) #PLSRegression模型
#regr = ElasticNetCV(cv=5, random_state=0) #ElasticNetCV模型
#regr = SVR(kernel="linear", C=100, gamma="auto") #SVR-linear模型
#regr = SVR(kernel="poly", C=100, gamma="auto") #SVR-poly模型
#regr = linear_model.LinearRegression() #线性模型
regr.fit(train_x, train_y.values.ravel()) #构建模型
y_pred = regr.predict(test_x) #预测表型
y_pred = pd.DataFrame(y_pred) #修改表型格式
test_all=test_y.reset_index() #修改验证表型的个体顺序
test_all['Pred_y']=y_pred #合并预测表型和验证表型
test_all.columns = ['taxa', 'Real_Y', 'Predicted_Y'] #添加表头
corr = test_all['Real_Y'].corr(test_all['Predicted_Y']) ##calculate correlation info ##预测数据和真实数据相关性
#coef = sm.OLS(test_all['Predicted_Y'], test_all['Real_Y']).fit().params[0] ##回归系数
data_results[i]=corr #保存结果
#保存数据
test_all=pd.DataFrame.from_dict(data_results, orient='index')
#导出csv格式
test_all.to_csv('alfalfa_100rep_ind_mean_SVR_poly_coefficient.csv', sep=',')
得到结果文件如下:

绘图
根据结果绘图。
library(ggplot2)
mydata<-read.table("combine_five_method_results.txt",header=TRUE,sep="\t")
mydata$Accuracy=abs(mydata$Accuracy) #相关系数有正负,准确率只考虑正值
p1<-ggplot(mydata, aes(x=Phenotype, y=Accuracy, fill=Method))+labs(x="Phenotype", y="Prediction accuracy") +
geom_boxplot(width=0.8)+ylim(0,1)+
theme_classic()+theme(
legend.position="right",legend.title = element_text(color = "black", size = 10), legend.text = element_text(color = "black", size = 10),axis.text = element_text(color="black",size=10),axis.title.x=element_text(margin = margin(t = 10, r = 0, b = 0, l = 0),size=10),axis.title.y=element_text(size=10))+
scale_fill_manual(values=c("#DD3226","#5EBCD5","#F88301","#07B294","#F1AC00"))
ggsave(plot=p1,"GS_accuracy.pdf", width = 18, height = 8, units = "cm")
其他结果
如果只是以上分析,显然不足以支撑起一篇文章。该文还探索了GWAS关联位点与GP全基因组标记的模型准确性的比较。从文献中总结了通过转录组学、蛋白质组学和数量性状位点定位的与FD相关的关键基因(但这些基因不能很好地预测紫花苜蓿FD)。
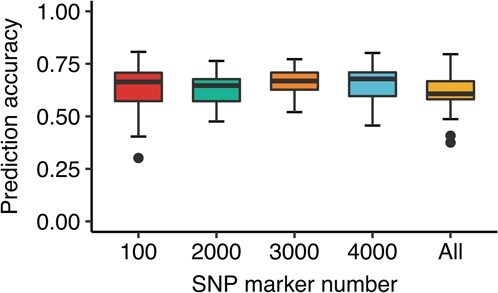
结果表明,使用具有线性核的SVM回归和前3000个GWAS相关标记对FD的预测准确率最高,为64.1%。对于植物再生高度,使用3000个GWAS相关标记和SVM线性模型预测准确率为59.0%。这比使用全基因组标记的结果(25.0%)要好。
总体来说,FD应该遗传力还算高,但预测精度并不高,这个不好评价,只是作为一个练习机器学习应用GP的例子。