新型冠状病毒SARS-CoV-2 S蛋白基因的变异与进化分析
食硕2201班 汪睿劼 22020080043 食硕2201班 张 琦 22020080063 食硕2201班 孙 允 22020080037 食硕2201班 韩秋雨 22020080043
摘要:新型冠状病毒SARS-CoV-2 引发的人类冠状病毒病COVID-19,仅仅2个多月时间在我国及国际上70多个国家出现迅速传播,致病和死亡率高,人类生命受到了极大威胁。一些科学家火速投入研究,对 SARS-CoV-2 的来源和进化、形态特征和基因结构、感染和致病分子机制开展深入研究。本实验主要利用生物信息学技术与方法,通过对SARS-CoV-2病毒与以蝙蝠、穿山甲为宿主的病毒S蛋白基因和氨基酸序列的比对分析,探究新型冠状病毒SARS-CoV-2的进化、变异情况,同时预测其S蛋白基因中插入序列的功能及对毒力的影响。
关键词:新型冠状病毒、序列对比分析、进化分析
1 引言
1.1新型冠状病毒SARS-CoV-2来源与进化
自2019年12月8日起,在中国武汉爆发了以呼吸道为主要症状的肺炎,经二代测序、病毒分离和鉴定等手段,确定为一种新型的冠状病毒[1]。世界卫生组织于2020 年1月19日将其暂命名为2019新型冠状病毒(2019 Novel Coronavrius, 2019nCoV), 于2020年2月11日正式命名为SARS-CoV-2病毒,引起的疾病称为2019冠状病毒病(COVID-19) [2]。截至北京时间2020年6月16日6时33分,全球新冠肺炎累计确诊病例超800万,达到8003021例;累计死亡病例超43万,达到435619例。
COVID-19确诊病例迅速增多,其传染性高,症状比较隐匿,世界卫生组织深感忧虑,人类生命受到极大威胁。国内外学者迅速对SARS-CoV-2 的来源、基因组及其结构、病毒分类、侵入和致病机制等进行研究并取得了重大进展,对防控 SARS-CoV-2 的蔓延和 COVID-19 的治疗及其药物研发产生了重大作用。
中国科学院武汉病毒研究所石正丽研究团队[1]根据从几位重症肺炎患者分离的病毒进行基因组测序,相应进行基因结构的生物信息学分析,发现该病毒全基因组序列与以前研究的 SARS 冠状病毒( SARS-CoV) [3]相似度高达 79.6%,与蝙蝠冠状病毒 SARSr-CoV 相似度高达 96% 。同时,从一名危重病人的支气管肺泡灌洗液中分离出的病毒可被几名肺炎患者的血清中和。复旦大学张永振研究团队和武汉市市中心医院合作研究,从一名武汉华南海鲜市场上工作的患有严重呼吸系统综合症患者中分离出新病毒,全基因组测序及其系统进化分析表明为一种新的冠状病毒科RNA 病毒,与一组来源于蝙蝠的SARS 样冠状病毒基因组核苷酸序列相似度高达 89.1%[4]。
香港大学袁国勇等多位学者的研究表明,冠状病毒中的α冠状病毒和β冠状病毒通常会导致人类呼吸系统疾病。根据目前对基因组序列数据库分析,蝙蝠和鸟类是冠状病毒基因源的理想寄主,促进冠状病毒进化和传播[5,6]。现发现的人类冠状病毒 SARS-CoV、中东呼吸综合症冠状病毒 MERS-CoV、两种新型人冠状病毒 HCoV-NL63 和 HCoV229E 均被认为来源于蝙蝠,蝙蝠很可能是α-冠状病毒和β-冠 状病毒的主要天然贮藏库,而啮齿动物和家畜可能作为中间宿主发挥重要作用,使病毒能够从自然宿主传播到人类。
上述研究结果表明,认为引起湖北武汉突发 COVID19的 SARS-CoV2 与 SARS 样冠状病毒有关,可能由蝙蝠 SARSr-CoV 演变而来,提示人类很有必要为未来 SARS 样疾病风险的出现做好应对准备。
1.2 新型冠状病毒 SARS-CoV2 的形态和基因结构
该病毒在分类学上属于冠状病毒科( Coronaviridae) 冠状病毒属( Coronavirus) 中的β类型冠状病毒,具有囊膜和刺突细胞学特征,基因组为线性单股正链的 RNA( ( + ) ssRNA) 病 毒。
全基因组测序结果展示该病毒基因组由29891个碱基组成,GC 含量为38%,共编码9860个氨基酸。与其他 βCoV 相似, 2019-nCoV 基因组包含两个位于侧翼的非编码区 ( 5'-UTR 和 3'-UTR) 、由 12 个开放阅读框( orf1ab, Spike, orf3a, orf3b, E,M, orf6, orf7a, orf7b, orf8 /orf8b, N 和 orf9b) 组成的一个编码多蛋白长开放阅读框、1个前导序列、9个转录调控序列组成。其基因组的排列顺序为 5'-复制酶( orf1 /ab) -结构蛋白[Spike( S) Envelope( E) -Membrane( M) -Nucleocapsid( N) ]3',其编码产物依次是复制酶( orf1 /ab) 、刺突糖蛋白( S) 、包膜蛋白( E) 、膜蛋白( M) 、核衣壳( N) 及其它假想蛋白,但缺乏在 β 类型冠状病毒 A 系中所特有的血凝素脂酶( HE) 基因。2019-nCoV 两侧的非编码区分别是由 265 和358 个碱基组成,与其它 β 类型冠状病毒两侧非编码区的相似度在83.6%以上。
已有对 SARS 的研究发现,该病毒在微观结构上具有包膜,包膜表面形成的刺突和膜上相应具有S蛋白和M蛋白。对S蛋白与宿主结合的受体结合域 ( receptor binding domain, RBD) 及其三维结构受到研究者高度重视。
1.3 生物信息学方法
NCBI (National Center for Biotechnology Information)是指美国国立生物技术信息中心,在生物学研究中,NCBI提供了大量的生物信息分析工具以及文献,它是美国国家医学图书馆(NLM)的一部分(该图书馆是美国国家卫生研究所的一部分),我们可以通过该信息库搜索所需要的各种生物学信息。Bioedit是一个序列编辑器与分析工具软件。功能包括:序列编辑、外挂分析程序、RNA分析、寻找特征序列、支持超过20000个序列的多序列文件、基本序列处理功能、质粒图绘制等等。MEGA的全称是Molecular Evolutionary Genetics Analysis,即分子进化遗传分析。MEGA可用于序列比对来、进化树的推断、估计分子进化速度、验证进化假说等。MEGA还可源以通过网络(NCBI)进行序列的比对和数据的搜索。pymol是一个开放源码、由使用者赞助的分子三维结构显示软件。pymol适用于创作高品质的小分子或是生物大分子(特别是蛋白质)的三维结构图像。
通过NCBI数据库及其所有的BLAST-N比对功能得到需要的核酸及氨基酸序列,接着进行聚类分析得出不同序列间的同源性及其核酸、蛋白质序列的差异。而后根据结果构建进化树得到不同病毒间的S蛋白序列远近关系。再通过蛋白质结构同源建模构建S蛋白模型,观察模型结果,研究并思考某些具体位点插入的位置,推测其可能的功能。
1.4 研究内容和意义
本实验主要利用生物信息学的方法,对SARS-CoV-2病毒的S蛋白进行分析,从NCBI中下载相关病毒全基因组序列,运用生物信息学软件MEGA-X、BEAST等软件,构建基因组进化树,推测病毒的时间进化信号,分析S蛋白的结构,探究新型冠状病毒SARS-CoV-2的进化、变异情况。
2 材料与方法
2.1 实验材料
从国家生物技术信息中心(NCBI:https://www.ncbi.nlm.nih.gov/genome/) 的核酸数据库中下载1株SARS-CoV-2病毒的全基因组序列文件和全基因组序列注释文件(Wuhan-Hu-1,RefSeq: NC_045512.2),而后通过NCBI的BLAST-N搜索比对其Spike蛋白(S蛋白)序列得到两株S蛋白序列相近的蝙蝠(GenBank: MN996532.1)、穿山甲(GenBank: MT121216.1)冠状病毒序列,并提取三者的S蛋白序列保存为FASTA格式。
2.2 实验方法
2.2.1 NCBI数据库检索SARS-CoV-2序列实验步骤
2.2.1.1 登录NCBI(https://www.ncbi.nlm.nih.gov/),在首页输入框内键入“SARS-CoV-2”,按search按钮;
2.2.1.2 在跳出来的输入结果中点击“Genomes”部分的“Genome”项,点击右侧数字一进入;
2.2.1.3 在网页下面“Representative(genome information for reference and representative genomes)”中的RefSeq栏中有编号 NC_045512.2,点击编号链接进入Wuhan-Hu-1序列的Genbank格式页面;
2.2.1.4 在GenBank格式网页上搜索”Spike”(可按Ctrl+F键),找到S蛋白的CDS序列,点击CDS链接,在跳出框中再点击“Display:FASTA”的链接,打开Spike基因的序列网页。
2.2.1.5 点击页面右上角的“Send to”,在下拉菜单中选择“Complete Record”,Destination选择“File”,Format选择“FASTA”,保存到工作目录。
2.2.1.6 再下载Spike蛋白的氨基酸序列。
2.2.2 BLAST搜索同源序列实验步骤
2.2.2.1 选择NCBI首页右侧的BLAST,选择nucleotide (blastn) 模式,upload刚才下载的Spike基因序列,根据自己的要求调整search set(一般默认),点击BLAST按钮;
2.2.2.2 在BLAST结果中点击Accession number下载需要的序列。
2.2.2.3 选择NCBI首页右侧的BLAST,选择protein (blastp) 模式,upload刚才下载的Spike基因序列,根据自己的要求调整search set,参数设置如下:数据库选择:非冗余蛋白质数据库,non-redundant protein sequence (nr);生物:病毒,viruses(taxid: 10239;比对算法:blastp,点击BLAST按钮;
2.2.2.4 在BLAST结果中点击Accession number下载蝙蝠(bat)、穿山甲(pangolin)等宿主的病毒序列。
2.2.3 SARS-CoV-2多序列比对实验步骤
2.2.3.1 将下载好的Wuhan-Hu-1、bat、pangolin的S蛋白序列和基因序列分别合并到一个文件(FASTA格式);
2.2.3.2 用bioedit打开文件,按Ctrl+A选中全部序列,选择“Accessory Application->ClustalW Multiple alignment->Run ClustalW”;
2.2.3.3 检查比对结果,观察人SARS-CoV-2与蝙蝠、穿山甲病毒S基因与蛋白质序列的差异,有多少核苷酸变异,有多少氨基酸变异,有哪些插入缺失(indel)突变,并保存结果。
2.2.4 进化树构建实验步骤
2.2.4.1 利用MEGA构建进化树,在mega中打开比对好的fasta文件,选择“analyse”;
2.2.4.2 选择“PHYLOGENY -> construct/Test Maximum Likelihood Tree…”
2.2.4.3 根据需求选择参数、是否进行bootstrap检验等。为加快建树速度,bootstrap值先设置为100,一般正式研究要求做1000次重复。
2.2.4.4 根据进化树结果,讨论来自不同地区的新冠病毒之间的进化关系,新冠病毒与蝙蝠、穿山甲病毒的进化关系。
2.2.5 蛋白质结构同源建模实验步骤
2.2.5.1 在Wuhan-Hu-1序列的Genbank页面搜索(Crtl+F)S蛋白,将氨基酸序列复制到文本文档中,命名protein.fasta;
2.2.5.2 浏览器打开Swiss-model网站(https://swissmodel.expasy.org),点击start modeling;
2.2.5.3 上传protein.fasta,点击Build Model,开始寻找模版并建模,也可以手动“search for template”,挑选高质量模版进行建模。
2.2.5.4 建模完成后,点击Model_01下载pdb模型文件。
2.2.5.5 在pymol等软件中打开下载好的pdb文件,点击软件底部的“S”(黄框)可以显示模型蛋白质的氨基酸序列,用鼠标拖动选取的序列会在3D模型中以小红点形式显示;
2.2.5.6 观察SARS-CoV-2的S蛋白的插入序列“PRRA”所处的位置,及思考此插入序列的可能功能。
3 结果
3.1 NCBI数据库检索SARS-CoV-2序列实验结果
3.1.1 SARS-CoV-2 S蛋白基因序列数据及详解
结果见图3-1-1。

3 1 1 SARS-CoV-2 S蛋白基因序列
3.1.2 SARS-CoV-2 S蛋白氨基酸序列数据及详解
下载Spike蛋白的氨基酸序列的方法为:在Wuhan-Hu-1序列的Genbank页面搜索(Crtl+F)S蛋白,将氨基酸序列复制到文本文档中,命名protein.fasta。
结果见图3-1-2。

3 1 2 SARS-CoV-2 S蛋白氨基酸序列
3.2 BLAST搜索同源序列实验结果
3.2.1 蝙蝠、穿山甲同源S蛋白核酸序列数据及详解
结果见图3-2-1-1和3-2-1-2。



3 2 1 1 蝙蝠同源S蛋白核酸序列



3 2 1 2穿山甲同源S蛋白核酸序列
3.2.2 蝙蝠、穿山甲同源S蛋白氨基酸序列数据及详解
结果见图3-2-2-1和3-2-2-2。



3 2 2 1 蝙蝠同源S蛋白氨基酸序列



3 2 2 2穿山甲同源S蛋白氨基酸序列
3.3 SARS-CoV-2多序列比对实验结果
3.3.1 SARS-CoV-2与蝙蝠、穿山甲S蛋白基因同源序列比对数据及详解
对比结果见图3-3-1-1。
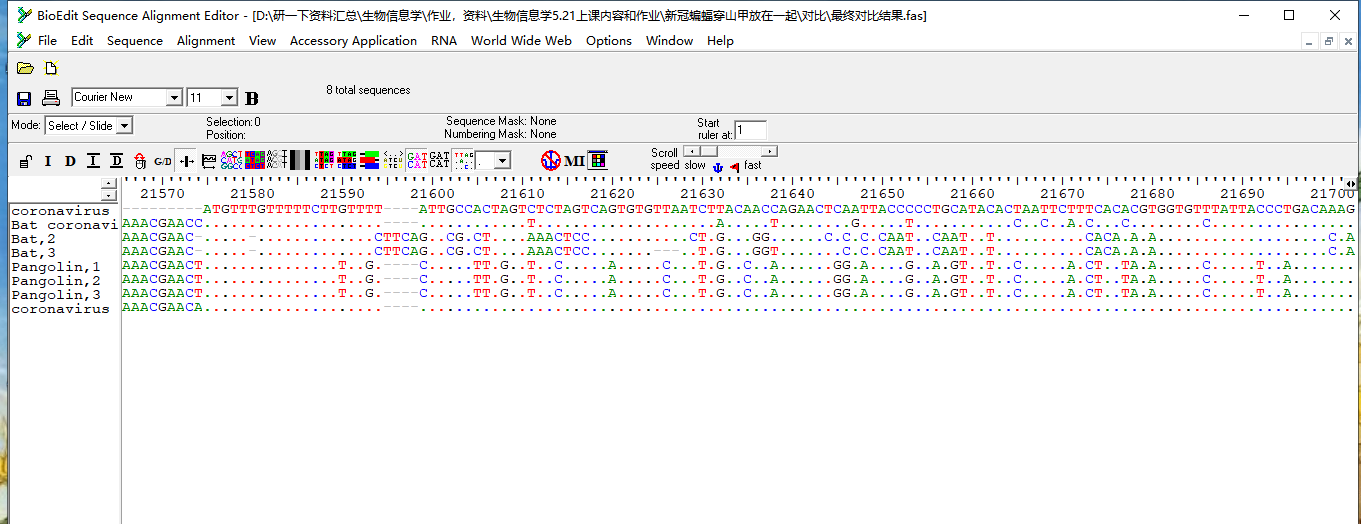
3 3 1 1 SARS-CoV-2与蝙蝠、穿山甲S蛋白基因同源序列比对
从基因层面来看, SARS-CoV-2 S蛋白基因与蝙蝠S蛋白基因和穿山甲S蛋白基因之间的差异分别为:当把穿山甲作为比较目标, SARS-CoV-2 S蛋白基因与其的相似性是84.5%,蝙蝠 S蛋白基因与其的相似性是88.5%[1]。
SARS-CoV-2 S蛋白基因有一处重要的插入突变,序列为TCCTCGGCGGGC,如图3-3-1-2所示。
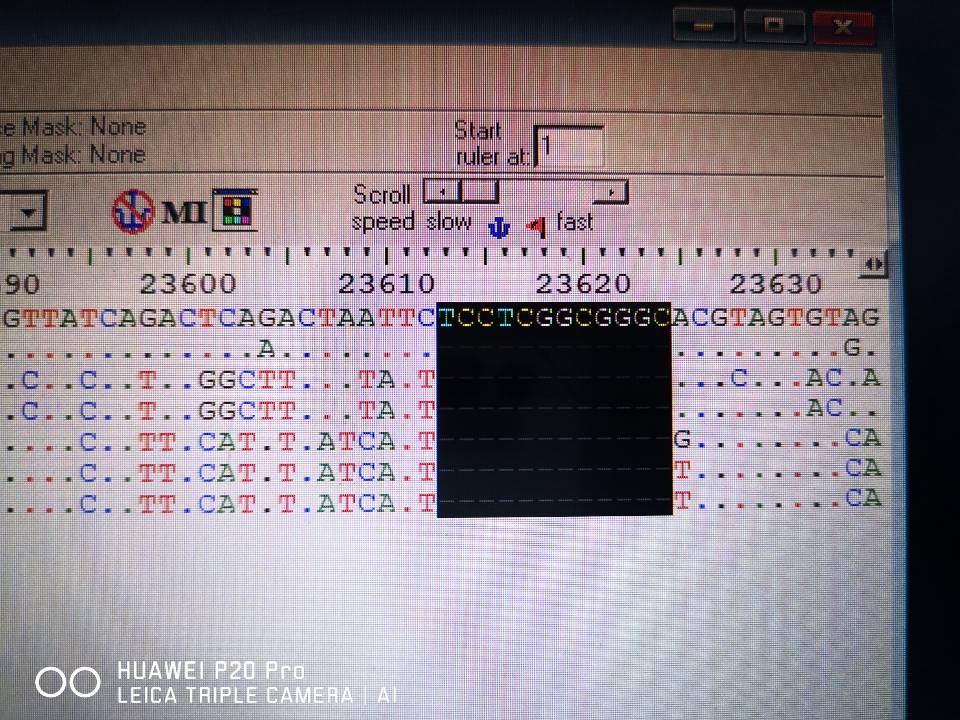
3 3 1 2 SARS-CoV-2 S蛋白基因的插入突变
3.3.2 SARS-CoV-2 S蛋白氨基酸与bat、pangolin同源序列比对数据及详解
对比结果见图3-3-2-1。
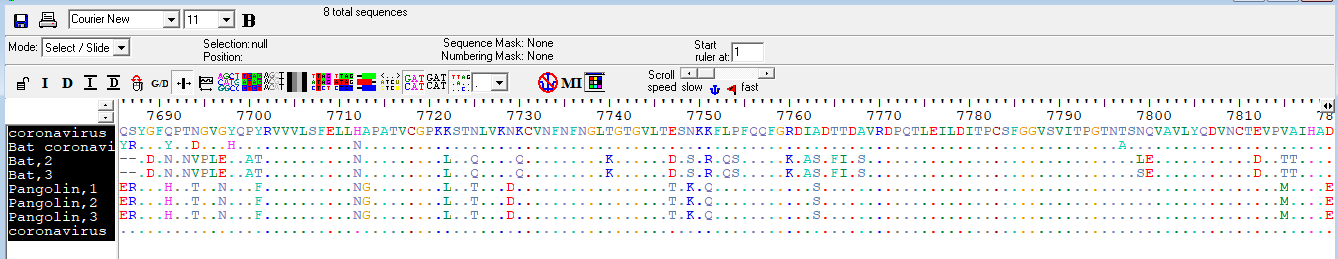
3 3 2 1 SARS-CoV-2 S蛋白氨基酸与bat、pangolin同源序列比对
从氨基酸层面来看, SARS-CoV-2 S蛋白基因与蝙蝠S蛋白基因和穿山甲S蛋白基因之间的差异分别为:当把穿山甲作为比较目标, SARS-CoV-2 S蛋白基因与其的相似性是90.7%,蝙蝠 S蛋白基因与其的相似性是89.8%[1]。
SARS-CoV-2 S蛋白氨基酸有一处重要的插入突变,序列为PRRA,如图3-3-2-2所示。
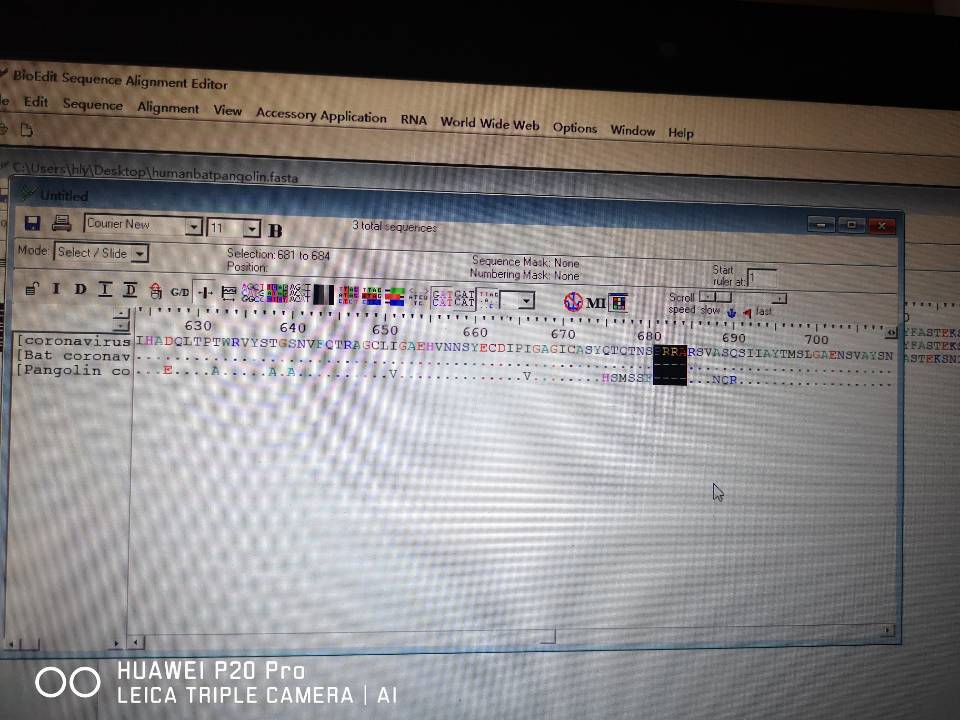
3 3 2 2 SARS-CoV-2 S蛋白氨基酸的插入突变
3.4 进化树构建实验结果
3.4.1 SARS-CoV-2与蝙蝠、穿山甲S蛋白基因同源序列进化树
构建结果如图3-4-1-1所示。
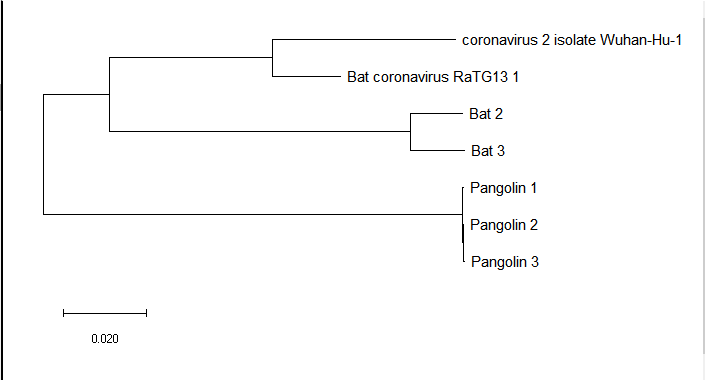
图3 4 1 1 进化树
不同地区的新冠病毒之间的进化关系如图3-4-1-2所示[2]。
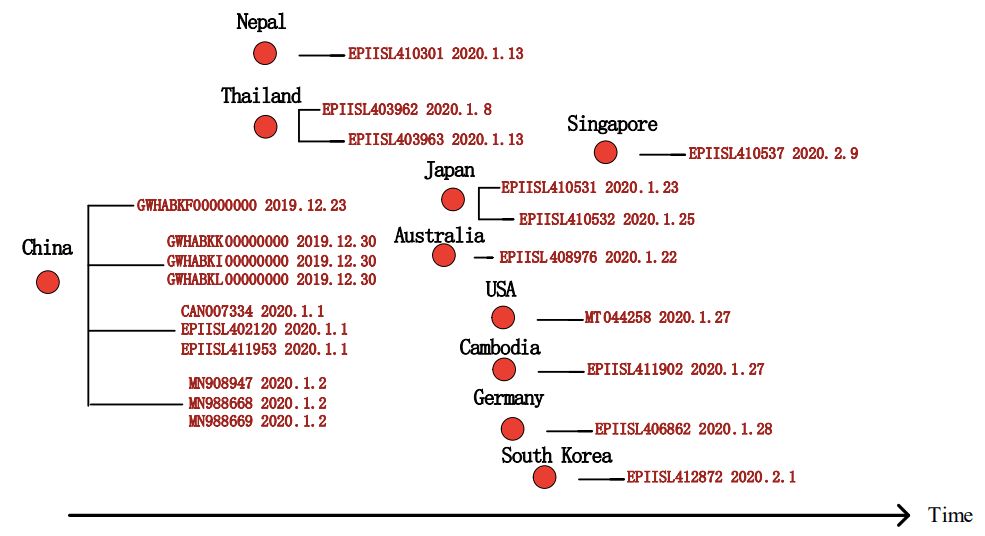
图3 4 1 2 不同地区的新冠病毒之间的进化关系
根据相关文献,新冠病毒可能源于穿山甲冠状病毒与蝙蝠冠状病毒的重组[1]。
3.5 蛋白质结构同源建模实验结果
3.5.1 蛋白质结构同源建模数据及详解
蛋白质结构同源建模结果如图3-5-1所示,其中红色部分为PRRA插入突变。
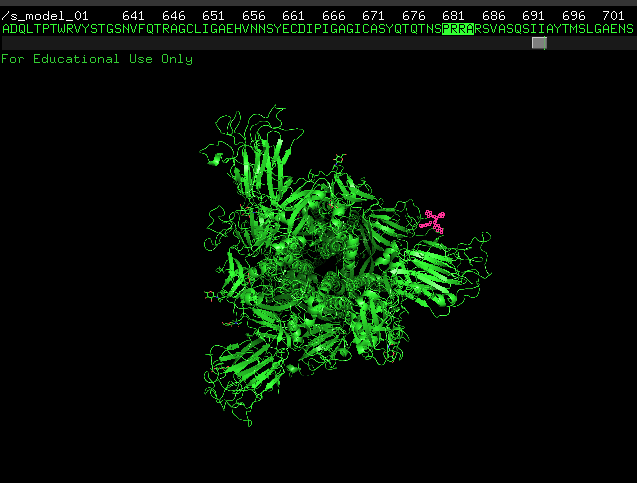
3 5 1 蛋白质结构同源建模
PRRA四个氨基酸的插入,从而形成能被人类弗林蛋白酶识别的“RRAR”序列。弗林蛋白质是切割某些冠状病毒和流感病毒S蛋白的常见酶,在各种组织和细胞中广泛分布,不仅在病毒感染的宿主表面可以发挥酶切效果,在病毒合成和成熟过程中都能发挥切割作用,能够显著提高病毒的感染性。
4 讨论
本研究提示新冠病毒S蛋白中弗林蛋白酶切位点的插入,可能是其强大传染能力的一个重要关键因素,而人类弗林蛋白酶则可能是抗新冠病毒治疗的新靶点,开发弗林蛋白酶抑制剂则可能对于防治新冠病毒疾病具有重要意义。
参考文献
[1] Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China[J]. Nature, 2020, 579(7798): 265-269.
[2] Zhou P, Yang X L, Wang X G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin[J]. nature, 2020, 579(7798): 270-273.
[3] Guarner J. Three emerging coronaviruses in two decades[ J ] . Am J ClinPathol,2020,aqaa029.
[4] Zhou P, Yang X L, Wang X G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin[J]. nature, 2020, 579(7798): 270-273.
[4] Ge X Y, Li J L, Yang X L, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor[J]. Nature, 2013, 503(7477): 535-538.
[5] Cui J, Li F, Shi Z L. Origin and evolution of pathogenic coronaviruses[J]. Nature Reviews Microbiology, 2019, 17(3): 181-192.
[6] Yu W B, Tang G D, Zhang L, et al. Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2/HCoV-19) using whole genomic data[J]. Zoological research, 2020, 41(3): 247.
组员贡献
食硕2201班 汪睿劼 22020080043:文献查阅、整理
食硕2201班 张 琦 22020080063:论文编写、修改
食硕2201班 孙 允 22020080037 论文编写、修改
食硕2201班 韩秋雨 22020080043 论文的整合、讨论后修改