图形泛基因组(图泛)以节点和路径的形式存储和展示物种的变异信息,关键作用是扩展线性参考基因组的坐标系统,以容纳更多的遗传多样性区域。本文首先总结了图泛构建方法,然后讨论了图泛基因结构和变异的注释,最后描述了现有的图泛规模和应用实例,包括与GWAS结合的图泛应用等。
首个完全测序的基因组是1977年的噬菌体φX174。截至目前,已有数千个基因组被组装。
2005年,泛基因组的概念被提出,最初在细菌中被定义为不同菌株中所有基因的集合。由于真核生物基因组通常包括大的基因间区域,真核生物的泛基因组通常被定义为物种中所有DNA序列的集合,包括存在于所有个体中的核心基因组和存在于某些个体中的可有可无的基因组部分。
目前构建泛基因组,通常采用homolog-based,map-to-pan或graph databse方法将一个物种的所有DNA序列组合成一个简单的序列集合。然而,由于线性泛基因组只是将PAVs添加到参考基因组,因此无法用于清晰地表征个体间的变异来源或精确定位PAVs。因此,提出了基于参考序列和变异序列之间关系的图形泛基因组,其中节点代表序列,边代表不同序列之间的连接。然后,泛基因组以图形格式存储,有效地将参考基因组与遗传变异联系起来。
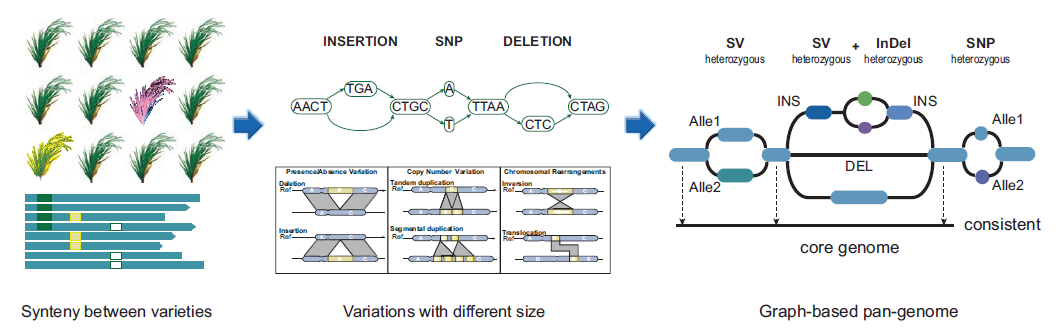
图泛构建方法可以分为两类:基于参考基因组和变异信息的方法,以及基于比对的方法。目前,用图形泛基因组替换线性基因组存在许多限制。图中经常缺少复杂的结构变异和基因组重排,但此方法弥补了线性泛基因组中SV位置的缺失。随着新工具和方法的出现,图形泛基因组将成为未来的新参考。
图形泛基因组的构建
图泛构建方法
基于图的泛基因组可以以图形式存储一个物种的所有遗传信息。一种方法是根据参考基因组识别基因组变异,然后将这些变异信息添加到线性基因组中。变异信息通常存储在VCF格式的文件中,其中可以包括小的变异或大的结构变异(>50 bp)。然而,VCF格式通常用于存储小的、简单的变异,不能正确表示嵌套或复杂的变异。
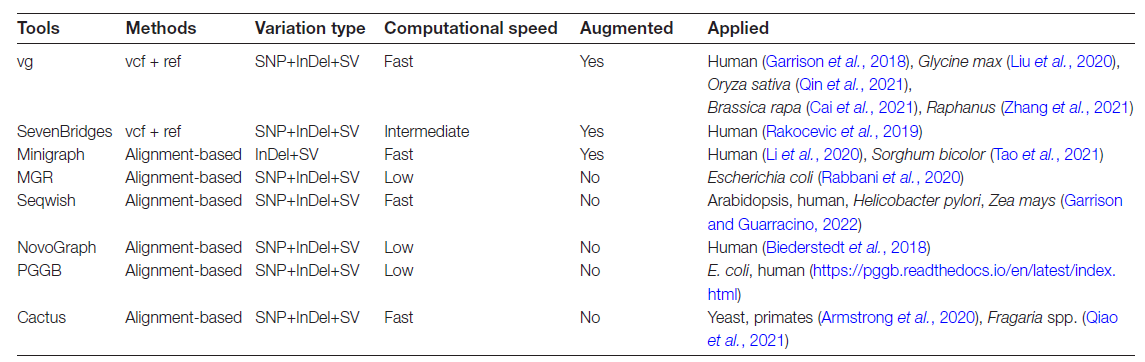
vg和Seven Bridges都使用VCF文件和参考基因组构建图泛,尽管vg还支持双向和循环图,导致不必要的计算复杂性,难以找到更多更完整变异信息的关键问题。两个解决思路:首先,在植物基因组中存在多次重复的情况下,现有的长读长比对工具通常在长的、近似相同的重复区域内产生错误的比对,而minimap2则通过使用对数间隙惩罚和查询特定的不匹配惩罚改善了重复区域中长InDels的灵敏性。其次,在SV鉴定过程中,可直接比较de novo基因组组装,或将reads比对到参考基因组。在辅助SV鉴定中选择Hi-C和Bionano也会导致最终变异信息的差异,从而影响图泛的质量。
此外,还有一种基于比对的方法可以通过将所有组装与参考基因组比对来直接构建图泛。MGR算法应用于多个大肠杆菌基因组,以构建第一个具有完整基因组的图泛。MGR算法压缩了输入基因组,使最终图泛比使用vg工具构建的要小。MGR图更适用于比较输入基因组之间的差异,而不适用作参考基因组。
minigraph方法应用于多个基因组,以构建一个具有最小contig长度≥100 kb的图泛,特别适用于染色体水平的基因组。这种方法比其他方法快得多,可以在3小时内构建包含20个人类基因组的图泛。minigraph构建的基于图的泛基因组只包含长度在50 bp到100 kb之间的SVs,SVs的数量多于dipcall发现的数量。然而,minigraph不能识别SNP,这可能会导致图中的偏差。
相比之下,由seqwish构建的图是无损的,并支持将PAF格式的all-versus-all比对结果转换为无损图。考虑到seqwish和vg都需要上游比对,PGGB和cactus提出了两种结合各种软件构建图泛的流程。PGGB主要使用wfmash、seqwish和smoothxg来比对成对序列并构建图泛,最后进行归一化以完成图的构建。此外,该流程还包括下游比对和可视化。Cactus主要使用minigraph和vg。首先,使用minigraph构建一个草图,然后将基因组与图对齐以添加小于50 bp的小变异。它还可以执行转换到vg格式并获取VCF格式的变异信息。此外,PGGB和cactus流程都建议单独为每个染色体构建图泛,因为处理整个基因组需要大量计算资源并复杂化下游分析。
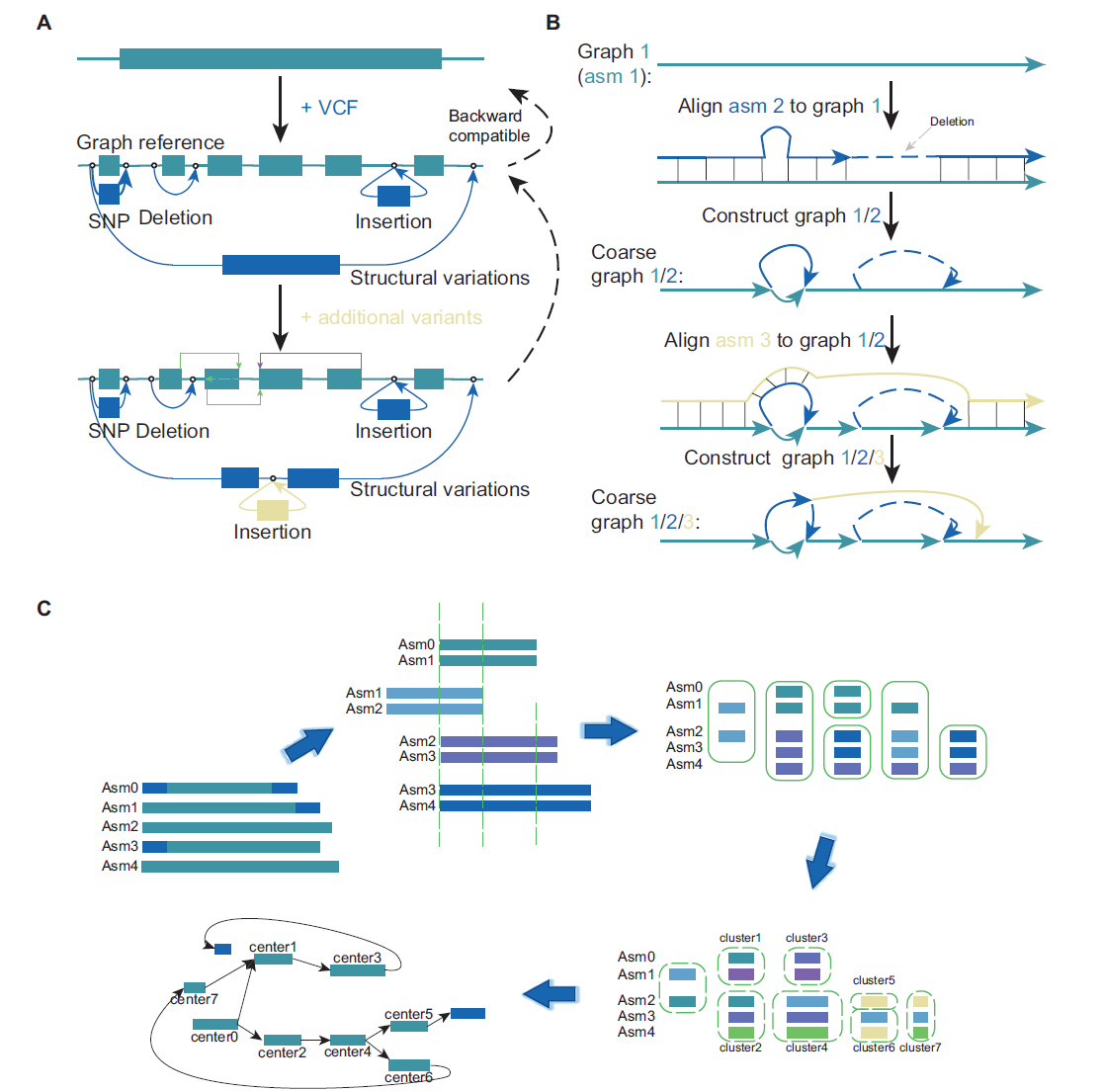
小编碎碎念:虽然植物泛基因组研究比人类报道更多,但方法学上仍是很肤浅的。今年(2023)3篇Nature论文连发公布了人类泛基因组首个草图,提出了Minigraph-Cactus新方法,可以作为动植物参考,尤其是高杂合度物种。
获取和合并PAV变异
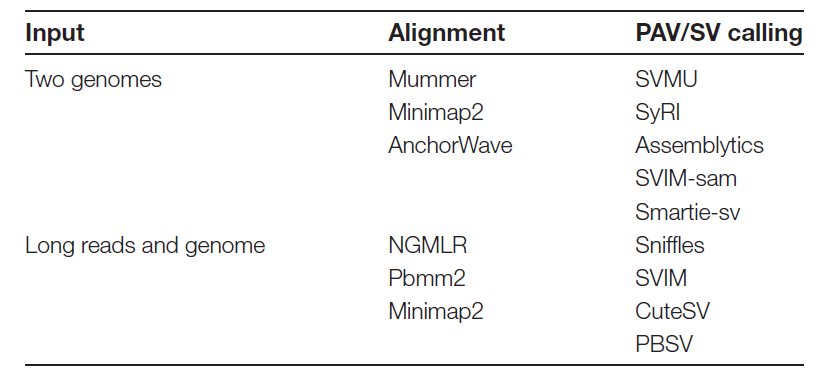
构建图泛的当前主流方法基于参考基因组和变异信息,在构建图泛之前需要准备参考基因组和其他基因组之间的变异信息(即获取PAVs)。 PAVs可以通过两种方式获得:通过基因组间比较或通过长读长与参考基因组的比对。MUMmer和minimap2是常用的基因组间比较工具。
由于植物中存在大量的转座子,一些针对脊椎动物开发的比对工具未能在复杂的植物基因组中产生预期的结果。最近开发的AnchorWave软件专为复杂的植物基因组而设计,它使用保守序列(如编码蛋白基因)作为锚定位点,以准确识别由转座子引起的基因组间差异。然后,根据比对结果执行PAV提取,并使用SVMU流程,SyRI流程,Assemblytics和SVIM-asm来提取SV信息。Smartie-sv是一个方便的工具,可以直接基于输入基因组输出SV信息。
许多比对工具是通过使用长读长和参考基因组开发的,包括NGMLR,minimap2和Pbmm2。Sniffles,SVIM和CuteSV可以从比对结果中提取SV信息。PBSV可以直接输入长读长和参考基因组以提取SV信息。考虑到存在冗余的PAVs,采用一种all-versus-all比较的方法对覆盖率大于50%的PAVs进行聚类。然后随机选择一个聚类中的PAV,并获得非冗余的PAV集合。这种方法简单且粗糙,因为同一聚类中的PAVs还包括SVs,随机选择其中一个作为代表会导致变异信息的丢失并在后续分析中产生错误。然后,过滤具有超过90%重复率的PAV序列,以获得最终的PAV序列。Survivor,Svimmer和Jasmine也可以用于合并冗余的PAVs。
图泛的存储格式
目前,图泛的存储和可视化是构建图泛的瓶颈。图泛由表示序列信息的节点和表示变异信息的边组成。
图形片段组装(GFA)可以存储序列图。GFA包括两种格式,v1和v2。每行的第一列是数据类型,GFA1中有八种数据类型:'#'表示注释;'H'表示标题;'S'表示节点(序列);'L'表示连接节点的边;'J'表示跳跃,用于定义与特定重叠或序列关联的段之间的连接(从v1.2版本添加);'C'表示包含,是两个段之间的重叠,其中一个包含在另一个内部;'P'表示参考基因组和单倍体的路径(Llamas等人,2019);'W'表示图中的有向路径(从v1.1版本添加)。S行表示参考基因组或单倍体,L行表示对应节点的名称和方向,可以更清晰地显示变异信息,更容易还原线性参考基因组的坐标,为基于图的泛基因组注释奠定了基础。W行适用于没有段之间重叠的图形(https://github.com/GFA-spec/GFA-spec/blob/master/GFA1.md)。
GFA2是GFA1的超集。与GFA1相比,主要区别如下:L行和C行被合并为E行;P行被替换为编码子图和路径的U行和O行;还有一个新的F行用于描述多重比对和一个新的G行用于描述脚手架。比对可以通过Dazzler-trace(https://dazzlerblog.wordpress.com/2015/11/05/trace-points/)或CIGAR字符串方法来描述,这是记录比对的两种方法(https://github.com/GFA-spec/GFA-spec/blob/master/GFA2.md)。
在minigraph方法中,基于GFA格式提出了参考GFA(rGFA)格式。基于GFA格式,只保留了段和链接数据,去除了节点之间的重叠,并在代表节点中添加了三个新标签:第四列表示染色体编号,第五列表示节点坐标,第六列表示节点来源。改进后的rGFA格式包含了在Hackathon活动中提出的图形坐标系的单倍体信息,以及与线性参考基因组相关的节点染色体编号和坐标。这种格式不仅可以还原坐标和完成线性参考基因组注释的迁移,还可以有效地提供路径、路径来源以及与路径相关的坐标,这极大地便于后续比较和比较信息的存储。
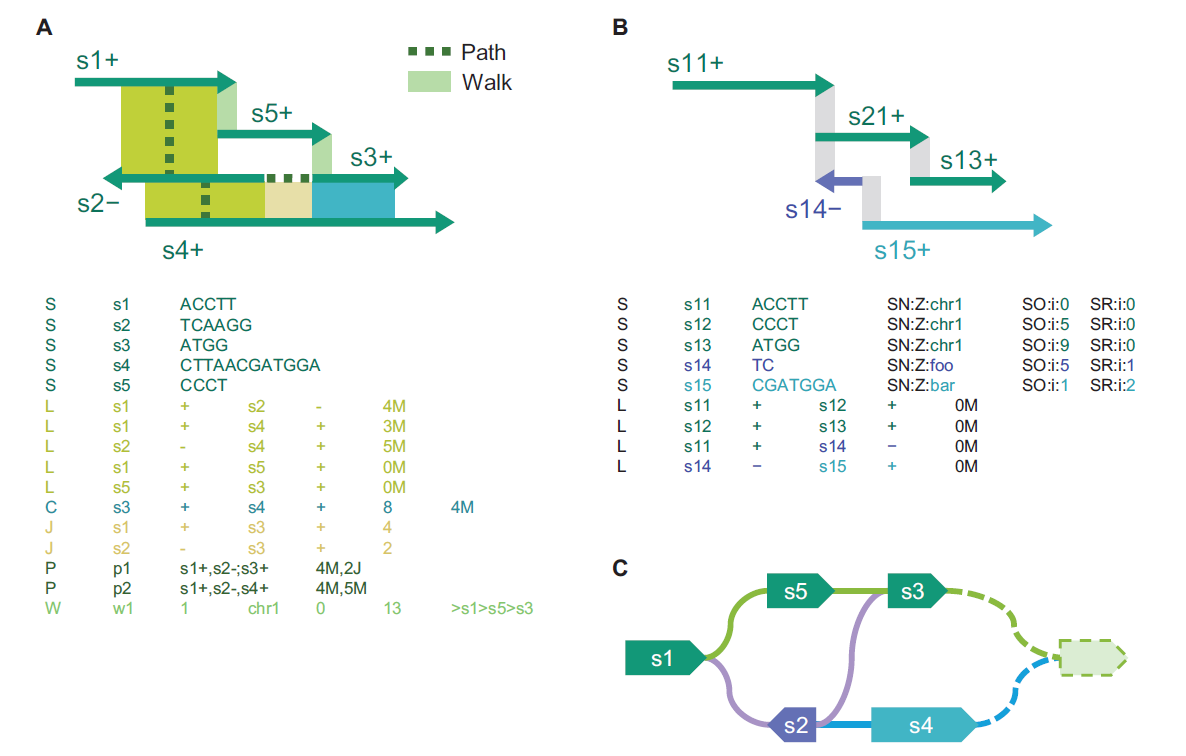
vg格式中还有一个二进制文件,可以与GFA格式相互转换。它通常用于下游分析,如vg映射和giraffe分析,用于构建索引文件或直接用作输入。ODGI是从GFA进行无损转换,并以二进制格式存储。这种格式具有与基于基因组序列的现有工具保持向后兼容性的优势。
线性参考基因组坐标的恢复
应用图泛的一个难点是与线性参考基因组坐标的对应关系。图泛不仅包含线性参考基因组,还包括大量的结构变异。
在表示图泛的rGFA格式中,虽然可以基于线性参考基因组标记图泛基因组节点的坐标,但由于图泛的复杂性,一些序列无法由线性参考基因组覆盖,这将导致节点坐标的丢失。此外,由vg表示的图泛允许节点序列出现在多个路径中,这使得图泛更广泛适用,但也增加了标记图泛节点坐标的难度,导致节点坐标的冲突。
对于来自非参考基因组的片段,坐标的恢复需要基于参考基因组坐标系统,由于在构建过程中的小变异过滤或变异发现中的假阳性/假阴性,目前无法准确表示源基因组中片段的坐标。因此,需要一个坐标系统来展现更好的可扩展性和可扩展性,以处理线性参考基因组未覆盖的区域,并随着线性参考基因组的更新,相应的图泛基因组同时更新节点、边和坐标。
图泛的可视化
目前,图泛的可视化方法主要分为两类:宏观水平方法(如Bandage、GfaViz和Panache)和基本水平方法(如vg viz、Sequence Tube Map和ODGI)。
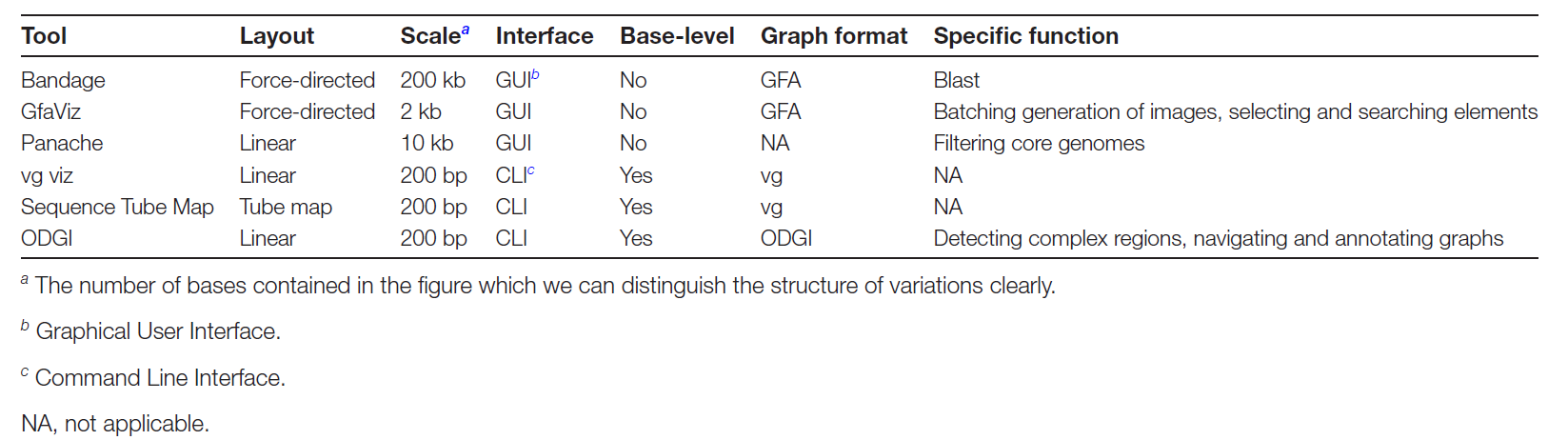
Bandage允许直接可视化GFA格式文件。除了一般可视化软件的缩放、旋转和着色功能外,Bandage还可以执行BLAST搜索并显示距离所选段固定距离的部分图形,这在可视化SVs时非常直观。
GfaViz支持两个GFA格式文件的输入,并支持两种布局算法。默认情况下,应力最小化算法将实现更好的可视化结果;对于较大的图形,用户可以选择快速多极多层方法算法。
Panache需要将GFA文件转换为bed格式作为输入,将图形泛基因组中的基因组划分为多个块,并根据块线性显示图形泛基因组;然后用户可以查看图形的特征,如核心基因组和可变基因组,并自定义核心和可变基因组的阈值以过滤核心基因组。
vg viz可视化生成的图形与Bandage获得的图形相似,但在基本水平上,构建图泛涉及的原始数据被添加到图像底部,以更直观地可视化变异的来源。
Sequence Tube Map可用于在基本水平分析变异,其中输入文件需要由vg进行索引。其基因组图的可视布局侧重于最大化所选基因组路径的线性度。通过预处理数据并提出要观察的区域,可以快速完成复杂多态性和SV的可视化和分析。
ODGI的功能非常强大,它可以根据不同需求重建图形并对其进行编辑,例如提取或连接感兴趣的区域,解开和导航泛基因组,以及对泛基因组图的排序和获取度量。
这两种方法各有优缺点。宏观水平更直观,构建速度更快,所需存储空间更小。可以在染色体级别观察SVs。尽管基本水平只能显示部分图形,但它包含的信息更丰富、更准确,因此认为结合这两种方法将使可视化更有用。
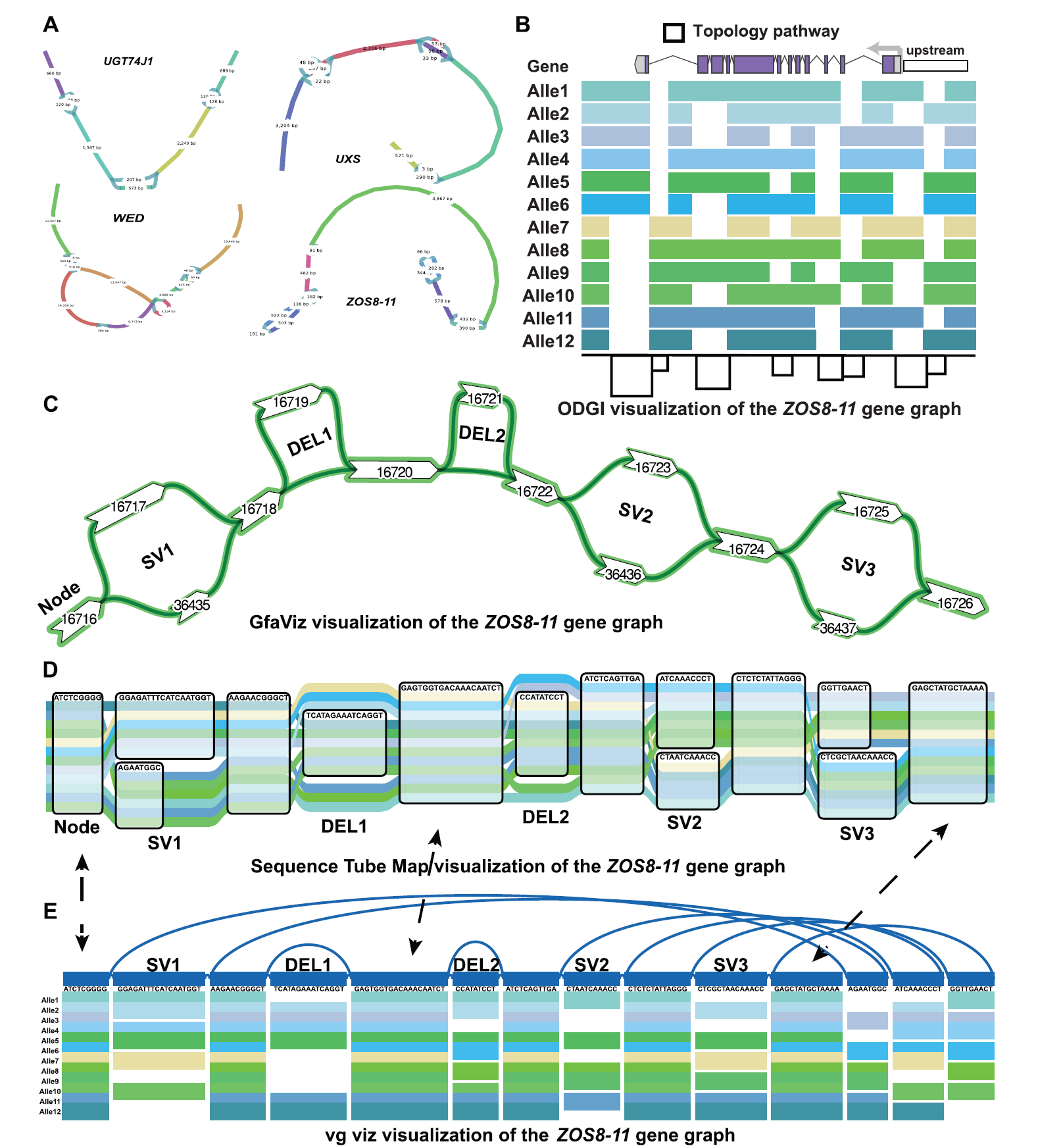
图泛的注释
基因结构注释
基因结构注释揭示了基因在基因组中的位置。在简单线性基因组中,已经建立了基于mRNA、表达序列标签和蛋白质序列的基因结构注释过程,通常通过直接或间接方式获得注释,随后应用统计模型和算法来预测新基因,这严格依赖于基因组坐标。
特殊的图形结构使得整个图泛的注释非常困难。图泛的路径可以有效替代线性基因组的坐标,从而可以直接将线性基因组中已知的基因映射到图泛上。vg软件直接将线性基因组从gff3格式转换为gGFF格式文件,其中基因组区间被完全替换为子图。
然而,基于线性基因组注释的这种映射只能映射已知存在于物种中的基因,不能预测新基因。未来的努力将致力于开发一组适用于图泛的算法,不仅可以直接映射线性基因组的原始基因,还可以直接预测可能存在于图泛中的新基因,这需要遍历图泛中的每个路径,并开发准确的图泛比较方法。
图泛的变异注释
变异注释的目的是识别它们的来源和类型,有助于下游分析。在使用vg构建的图泛中,内置工具可以用于完成变异注释。vg方法要求使用与线性参考基因组相同的坐标,因此变异位置也是基于相同的坐标集,允许直接使用线性参考基因组的注释文件。因此,rGFA文件提供了一组新的坐标。
未来可能需要通过不同的方法多次注释图泛,根据实际需要过滤注释,最终将所有注释合并成一个文件,形成最终的注释文件。
此外,在植物基因组中广泛存在大的倒位和易位,它们与育种密切相关。在这些SVs中,尽管序列没有改变,但位置发生了改变,不管是否有注释,这都不会影响后续比较中的变异识别。但是,如果在序列比对中发现相同的结构变异并提前进行注释,对于后续分析非常方便。
导致SVs的驱动因素之一的可转座元件(TEs)与作物中的许多表型相关,因此TEs的注释也应予考虑。TEmarker利用参考基因组、其TE库和短序列来创建泛基因组TEs。这个工具还可用于TE的基因型和基于TE的GWAS,为研究TE驱动的结构变异提供新的见解。
结构变异鉴定与基因分型
图泛比对软件的优势
图泛可以用作在更广泛的群体中调用结构变异(SV)和基因型的新参考基因组。
目前,线性基因组比对工具难以应用于图泛比对。已经开发的基于图的比对算法和软件,包括vg map、GraphAligner、Hisat2、V-MAP和vg giraffe。
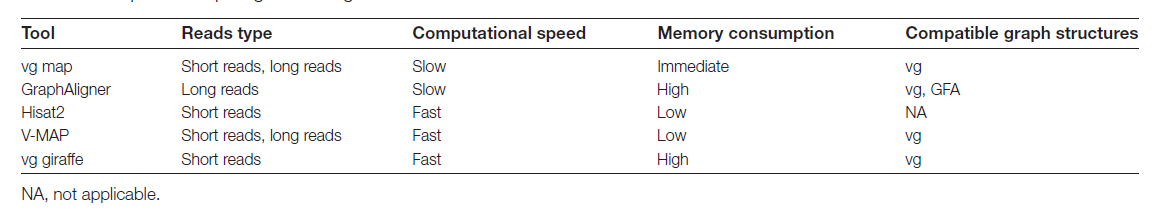
GraphAligner将两种序列对序列比对算法(Shift–And算法和Myers的位向量算法)扩展到能处理任意图形中。
Hisat2是一种低内存消耗、快速比对的软件,采用了图Ferragina–Manzini索引。它不需要事先构建基于图的泛基因组。
V-MAP可以有效地识别基因组图的小子图,以实现最佳读数比对。
Giraffe是一种具有相对高速度和准确性的短读比对工具,能够比对复杂的图形区域;目前是图泛短读长比对的最佳工具。
基于上述比对结果,可以进行SV基因型分型和新的SV鉴定。在使用短读长进行SV基因型分型时,vg call和paragraph都优于传统的SV基因型分型工具,PanSVR可以提高人类短读长数据的比对质量和SV基因型分型。
应用群体特异的图泛进行序列比对,可以在各个方面提高结果比对的质量,还可以在变异基因型分型期间发现更多的SNP、InDel和SV。SV的长度也明显大于线性基因组。使用vg augment和vg call可以扩展图,构建新的图并鉴定新的SV。
除了在人类泛基因组中实现的性能外,大豆、水稻和油菜等作物中已经完成了跨群体的SV基因分型,之后基于重测序数据恢复了与变异相关的一些表型信息,展示了图泛的优势。然而,在构建高质量的图泛之前,根据图泛的注释,判断重测序数据与图泛之间的比对是否准确仍然具有挑战性。
图泛的应用
功能元件的鉴定
基因表达受编码区域以外的非编码区域的影响。Cis调控元件是存在于非编码区域的一类重要调控元件,它们影响基因表达。位于Cis调控元件中的SV可能通过控制调控区域的变异来影响基因表达,从而改变物种的性状。
在番茄中,通过对100个番茄品种进行长读测序,发现了238,490个SVs,发现大约50%的SVs位于基因区域或基因上游调控区域(距编码序列±5 kb)。在100个基因组中,大约95%的基因的上游调控区域至少存在一个SV,其中大部分位于Cis调控区域。在对21,156个SV-基因对进行评估时,发现位于调控区域的1,534个SVs会显著影响基因表达。
尽管大多数影响基因表达的SVs位于基因区域,但也有一小部分位于Cis调控区域的SVs可能会影响基因表达。在玉米中,通过对368个玉米转录组数据和多态性SVs进行联合表达定量性状位点分析,发现一个长为1794 bp的SV作为Zm00015a037064基因的Cis表达定量性状位点,并通过影响相互作用来影响染色质,从而影响基因表达。
转座元件(TEs)是影响植物基因表达的重要组成部分,它们通过插入到基因组中的不同位置来影响基因表达,已知TEs通过插入到基因上游的启动子或增强子中来调控基因表达。通过比较和分析可访问染色质区域(ACRs)中的TEs,发现TEs的插入会影响ACRs中的甲基化状态,TEs的插入会影响附近基因的表达,从而证实TEs作为Cis调控元件调控基因。在水稻、番茄、油菜、萝卜、拟南芥中均有研究报道。
目前,构建图泛的主要方法是将线性参考基因组与PAV信息相结合。寻找具有更完整种群的PAVs可以使图泛更加稳健,特别是挖掘HOT区域。这些区域通常包含大量新基因,与抗性基因更相关,有助于育种改良。
现有物种中的图泛规模
植物图泛研究:大豆、油菜、高粱、水稻、萝卜、番茄、面包小麦、白菜、大麦、柑橘、牧草、黄瓜等。
基于图泛的GWAS
与SNP GWAS互补,在水稻、大豆、油菜中均有研究。
未来应用
图泛目前难以应用于复杂度高、基因组庞大的物种,而且尚未建立用于评估图泛的标准。构建图泛的目的是更清晰、更直观地了解物种的遗传多样性。然而,目前的大多数生物信息分析工具只适用于线性参考基因组。基于图的泛基因组的下游分析需要开发更多的算法和工具。除了挖掘新的SVs和与SV相关的表型特征外,还应开发更广泛的图泛应用,这些应用可以与多组学数据(蛋白质组数据、代谢组学数据)结合,进行多维关联分析,以识别候选位点,同时还应用于表观遗传学,例如将DNA甲基化和其他信息存储在图泛中,用于比较由不同等位基因的甲基化引起的不同表型。
结论与未来展望
随着测序成本的降低和第三代测序技术的发展,目前可以通过不同的方法构建图泛。与线性基因组相比,基于图的泛基因组包含更多的序列和变异,可以允许更多的reads与图进行比对。通过变异的注释,可以在基因组中恢复更多与变异相关的表型信息。因此,作物育种和改良的指导原则将从单一参考基因组转变为多参考基因组。
T2T基因组可作为参考基因组来构建更全面和更准确的图泛,有助于研究复杂区域和重复元件的遗传多态性,如着丝粒区域。目前,基于图的泛基因组学仍处于起步阶段,用户友好的在线资源稀缺。有必要构建一个植物泛基因组数据库,并开发与植物基因组特点相适应的构建方法,而不是直接使用用于构建人类图泛的软件。图泛将成为线性基因组的补充,而不是完全取代它们,这是由于线性基因组的成熟工具流程和用户习惯等因素决定的。将额外的线性基因组分析方法和工具迁移到图泛或将这两种方法结合使用将是一个主要挑战。
图泛也存在一些局限性。首先,目前还没有一种方法来评估从多个线性基因组构建的图泛质量。因为一个物种包括各种各样的品种,需要图泛具有高度的可塑性,并且能够有效地将新测序的基因组与现有的图泛整合在一起,即使它们在测序深度和测序方法上有所不同,也需要评估图泛是否能够有效地代表一个物种。这不仅限制了物种基因组特征的分析,也限制了图泛在不同物种之间的比较分析。其次,当前可用的工具之间的通用性较差,每种类型的软件都有其优点和缺点,无法相互补充。因此,完成所有上游和下游分析是困难的。另一个问题是图泛的可视化,特别是有关图形结构的清晰可视化,以便更容易理解图的拓扑结构和变异信息。
有一种新趋势是将图泛作为新的参考基因组来应用,它比线性基因组更大更完整,包含了几乎所有物种的基因和变异信息。minigraph、vg等工具可构建图泛的完整工作流程,允许快速构建图泛和下游分析。
本文整理自广西大学宋佳明和陈玲玲老师(原华中农业大学教授)的综述文章:Wang S, Qian YQ, Zhao RP, Chen LL, Song JM. Graph-based pan-genomes: increased opportunities in plant genomics. J Exp Bot. 2023;74(1):24-39. doi:10.1093/jxb/erac412