GWAS
GWAS + 选择进化 代码
library(CMplot) library(tidyverse) fst = choose.files() pi = choose.files() fst1 = read.table(fst, header = T) head(fst1) fst2 = fst1 %>% select(1,2,3 ......
GWAS:plink进行meta分析
之前教程提到过Metal是可以做Meta分析,除了Metal,PLINK也可以进行Meta分析。 命令如下所示: plink --meta-analysis gwas1.plink gwas2.plink gwas3.plink + logscale qt --meta-analysis-snp-f ......
GWAS数据库
NHGRI-EBI GWAS数据库: https://www.ebi.ac.uk/gwas/ 描述:由美国国家人类基因组研究所(NHGRI)和欧洲生物信息研究所(EBI)合作建立的GWAS数据库,提供了公开可访问的GWAS关联结果和相关信息。 GRASP: http://grasp.nhlbi.ni ......
GWAS:表型的标准化(the normalization of phenotype)
GWAS表型的标准化方法一般有Quantile normalization、Inverse rank normalization、Z-score normalization等。 各自区别如下: ## 一、Quantile normalization 该方法将每个样本中表型值进行排序,然后将其规范化到 ......
post-GWAS: transcriptome-wide association studies (TWAS) 结果解读
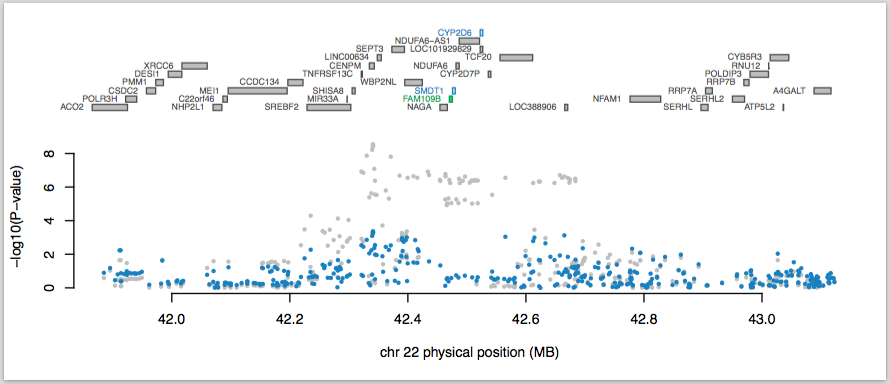 The top panel shows all of the genes in the locus. The marg ......
R语言实现GWAS结果显著SNP位点归类提取与变异类型转化
GWAS结果显著SNP位点归类提取与变异类型转化 根据GWAS得到的Rresult文件信息,能够找出每个snp位点对应的显著性情况和基因变异信息,接下来,需要根据表格中的信息进行归纳总结,对不同显著性层次进行区分,找出可能性最大的点,过程比较繁琐。 这里笔者分享一个算法,使统计SNP和变异类型变的更 ......